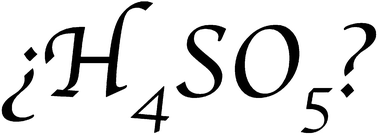Publications






350. Alvaro Muñoz-Castro. Through-Space Magnetic Response of Noble-gases. Phys. Chem. Chem. Phys. 2026. DOI: 10.1039/D5CP03897A
349. Margot Paco-Chipana, Alvaro Muñoz-Castro. Copper and Silver Cubooctahedron Cavities in Halide Encapsulation. Evaluation of Interaction Energy, and Optical Properties given by the Inclusion of Halides as Non-Innocent Templates. Inorg. Chem. 2026.
348. Peter L. Rodriguez-Kessler, Alvaro Muñoz-Castro Cyclo[48]carbon. Evaluation of its Inherent Magnetic Behavior and Anisotropy from DFT Calculations. Chemistry 2025, 7, 192.
347. Jonathan Fagan, Nabiha Syed, Wangshu Wen, Hanna Morales Hernández, Alvaro Muñoz-Castro, Christopher Johnson. Sequential Ag doping of Au25- atomically precise nanoclusters induces alternating positive and negative shifts of HOMO-LUMO gap. Chem. Sci.
DOI: 10.1039/D5SC06264K
346. Ya-Shan Huang, Ziqi Deng, Wen-Juan Tian, Hui-Qing Sun, Alvaro Muñoz-Castro, Teng-Teng Chen, Zhong-Ming Sun. σ-Aromatic Ge–Sn Bridges in a Triply-Bonded Bare Cluster Dimer with a 178-μs Long-Lived Triplet State. J. Am. Chem. Soc. 2025, 147, 45723-45730.
DOI: 10.1021/jacs.5c18030
345. Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. From Cationic to Anionic Al13 Clusters. Aromatic Characteristics for Intermediate Superatomic Species. J. Chem. Phys. 2025, 163, 224308. DOI: 10.1063/5.0300566
344. Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Linear vs Star-Shaped Be5O62- Cluster Arrays. Balance Between Repulsion and Electronic Stabilizing Terms. J. Nano. Res. 2025, 27, 314.
DOI: 10.1007/s11051-025-06506-9
343. Raúl Guajardo-Maturana, Macarena Rojas-Poblete, Plinio Cantero-Lopez and Alvaro Muñoz-Castro. Nature Interaction in CB[8]TPA 2 Ternary Sandwich Systems. Insights of the Host-Guest Binding into the Emission Features. Dyes and Pigments, 2026, 245, 113214.
DOI: 10.1016/j.dyepig.2025.113214
342. Mukundam Vanga, Benjamin T. Diroll, Alvaro Muñoz-Castro, Pavel Mykhailiuk, H. V. Rasika Dias. Trinuclear copper(I) and silver(I) complexes of a pentafluorosulfanyl-decorated pyrazolate. Inorg. Chem. 2025, 64, 25065-25076.
DOI: 10.1021/acs.inorgchem.5c03702
341. Vo Quang Huy Phan, Mukundam Vanga, Benjamin T. Diroll, Alvaro Muñoz-Castro, H. V. Rasika Dias. Gold chains and copper gyrobifastigia: trinuclear gold(I) and octanuclear copper(I) complexes derived from a fluorinated pyridyl ligand. Dalton Trans., 2025, 54, 15717-15725.
DOI: 10.1039/D5DT02150B
340. Dinesh Acharya, Hao Liang, Alvaro Muñoz-Castro, Di Sun. Optical and excited state properties of Au30(S-Adm)18 and Au38S2(S-Adm)20 nanoclusters: Accuracy from TD-DFT and TDDFT + TB approaches. J. Chem. Phys. 2025, 163, 164301.
DOI: 10.1063/5.0292988
339. Margot Paco-Chipana, Alvaro Muñoz-Castro. Impact in Increasing the Bent Angle in Nickel Porphyrin(2.1.2.1) Bows. Evaluation of Structural and Molecular Features From Computations. RSC Adv. 2025, 15, 42229-42237.
DOI: 10.1039/D5RA07496G
338. Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Consecutive Planar and Spherical Aromatic Arrays. Shielding Cone Behavior in Multiple Planar-Spherical Aromatics from Phenyl and Carboranes Motifs. Adv. Theory Simul. 2026, 9, e01293.
337. Samantha Ortega-Flores, Peter L. Rodríguez-Kessler, and Alvaro Muñoz-Castro. Size-Dependent Structural Motifs in AgnMo (n = 2–13) Clusters: From Planar to Icosahedral Architectures. J. Nano. Res. 2025, 27, 277.
DOI: 10.1007/s11051-025-06471-3
336. Margot Paco-Chipana, Peter L. Rodriguez-Kessler, and Alvaro Muñoz-Castro. Embedding a Guest Gold Cluster in an Organic Host. Evaluation of the Encapsulation Nature in Au18-Superphane Host-Guest Aggregate. Phys. Chem. Chem. Phys. 2025,27, 21544-21552
DOI: 10.1039/D5CP01989C
335. Jose Manuel Guevara-Vela, J. L. Cabellos-Quiroz, Sebastián Salazar-Colores, Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Structure search for B7Mn2 clusters: High-spin inverse sandwich structure. Comput. Theor. Chem. 2025, 1254,11548.
DOI: 10.1016/j.comptc.2025.115487
334. Rational Design of Antimony-Based Zintl Clusters: Unveiling Unconventional Bonding and Architectures. Yi Lai, Wei-Xing Chen, Wen-Juan Tian, Alvaro Muñoz-Castro, Gernot Frenking, Zhong-Ming Sun. Acc. Chem. Res. 2025, 58, 18, 2875-2885.
DOI: 10.1021/acs.accounts.5c00460
333. Peter L. Rodriguez-Kessler, Alvaro Muñoz-Castro. Cadmium Mediated Aggregation of Spherical-Planar and Spherical-Spherical Aromatic Lead-Based Clusters Towards Extended Cluster Materials. Chem. Phys. Lett. 2025, 880, 142394. DOI: 10.1016/j.cplett.2025.142394
332. Raul Guajardo-Maturana, Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Ag16 clusters as Host for Halide Encapsulation. Evaluation of Structural, Interaction Energy, and Optical Properties given by the Inclusion of Non-Innocent Templates. Inorg. Chem. Comm. 2025, 181, 115331.
DOI: 10.1016/j.inoche.2025.115331
331. Manas R. Samantaray, Tin Lok Wong, Abhay kumar Mondal, Sara Pescetelli, Duu-Jong Lee, Alvaro Muñoz-Castro, Rustono Farady Mart, Chung-Yu Guan, Baomin Xu, Antonio Agresti, Hsien-Yi Hsu. Lead-free MaSnI3/Sb2S3 heterojunction solar cell with power conversion efficiency approaching 30%: A SCAPS-CD simulation study. Solar RRL, 2025, 9, e202500642.
330. Miquel Solà, Alvaro Muñoz-Castro. Aromaticity of All-Metal Clusters. Chem. Commun. 2025, 61, 14280-14291.
DOI: 10.1039/D5CC03842A
329. Peter L. Rodriguez-Kessler, Alvaro Muñoz-Castro. From Cationic to Anionic Cu7 and Ag7 Cluster. Variation of Aromatic Behavior in Intermediate Superatomic Species. Chem. Phys. Lett. 2025, 878, 142316. DOI: 10.1016/j.cplett.2025.142316
328. José Manuel Guevara-Vela, Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro, Tomás Rocha-Rinza. Electronic and structural properties of magnesium-doped platinum clusters: superatomic features of the MgPt9 complex. Phys. Chem. Chem. Phys., 2025, 27, 17800-17810.
DOI: 10.1039/D5CP02053K
327. Ricardo A. Murcia-Galan, Sandra M. Leal-Pinto, Martha V. Roa-Cordero, Alvaro Muñoz-Castro, Desmond MacLeod-Carey, Mario A. Macías, John J. Hurtado. 1,3-bis(1,2,4-triazol-1-yl)-propan-2-ol as ligand in Co(II) and Cu(II) complexes: Synthesis, DFT calculations, and improved antifungal activity. J. Mol. Struc. 2025, 1346, 143274.
DOI: 10.1016/j.molstruc.2025.143274
326. Yuvaraja Dibdalli, Desmond MacLeod-Carey, Gabriel Abarca, Cesar Morales-Verdejo, Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. On the Role of Small Copper Clusters in Catalyzing Ammonium Perchlorate Decomposition: Insights from Density Functional Theory. Chem. Phys. Lett. 2025, 877, 142266. DOI: 10.1016/j.cplett.2025.142266
325. Raul Guajardo-Maturana, Carlos Rivera, Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Coating the Fullerene. Evaluation of the Core-Shell Interaction Nature in a Saturated Exohedral Cuprofullerene. Phys. Chem. Chem. Phys. 2025, 24, 15723-15730.
DOI: 10.1039/D5CP00944H
324. Raul Guajardo-Maturana, Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Evaluation of Halide Encapsulation in a Cuboctahedron Cu12 Cage. Structural and Interaction Energy in a Highly Coordinated Cavity in the [{TpMo(μ3-S)4Cu3}4(μ12-X)]- (X= Cl-, Br-, I-) Species. Chem. Phys. Lett. 2025, 877, 142254. DOI: 10.1016/j.cplett.2025.142254
323. Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Al2B7. An Extension to the Inverse Sandwich B9 Cluster Featuring Lewis Acid Sites And Planar Aromaticity. Phys. Chem. Chem. Phys. 2025, 27, 12793-12800.
DOI: 10.1039/D5CP01078K
322. Jose Manuel Guevara-Vela, P. L. Rodríguez-Kessler, J.L. Cabellos-Quiroz, Tomas Rocha-Rinza, Alejandro Vasquez-Espinal, A. Munoz-Castro. Structure of BnCu0/−2 (n = 2-13) clusters: size dependent properties. J. Phys. Chem. A. 2025, 129, 22, 4855-4860. DOI: 10.1021/acs.jpca.4c08443
321. Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Aryl-Crowding in the Carborane Cage. Aromatic Characteristics in a Spherical Aromatic Unit Crowned by Aromatic Rings. Dalton Trans. 2025, 54, 9679-9688 DOI: 10.1039/D5DT00787A
320. Wei-Dan Si, Lu-Yang Xing, Alvaro Muñoz-Castro, Chengkai Zhang, Bao-Liang Han, Jian-Long Zhou, Zhi Wang, Chen-Ho Tung, Di Sun. Bioinspired phosphate homeostasis for atomically precise regulation of silver nanoclusters. National Science Review, 2025, 12, nwaf183.
DOI: 10.1093/nsr/nwaf183
319. Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Three-fold Aromatic Boranes. Spherical Aromaticity in borane ortho-carboranes as an Useful Trimer Node for Cluster-Based Architectures. Phys. Chem. Chem. Phys. 2025, 27, 11112-11118.
DOI: 10.1039/D5CP00867K
318. Renato P. Orenha, Ana L. O. Andrade, Renato G. Rocha, Alvaro Muñoz–Castro, Thiago F. Santos, Maurício J. Piotrowski, Giovanni F. Caramori, and Renato L. T. Parreira. Ionic Recognition Controlled by Conformational Change: A DFT Investigation. ACS Omega 2025, 10, 16, 16114-16122.
317. P. L. Rodríguez-Kessler, Alvaro Muñoz-Castro. A Bridge Between Silicon and Gold Structures. Resemblance Between W@Si162+ and W@Au12 Clusters. Chem. Phys. Lett. 2025, 869, 142042. DOI: 10.1016/j.cplett.2025.142042
316. P. L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Structure search for B7Cr2 clusters: Inverse sandwich structure for the global minimum. Polyhedron, 2025, 273, 117486.
DOI: 10.1016/j.poly.2025.117486
315. P. L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Macrocyclic meta-Carborane Hexamer. Evaluation of Aromatic Characteristics As a Cluster-Based Analog to Phenyl-Bridged Macrocyclic Structures. Phys. Chem. Chem. Phys. 2025, 27, 6744-6750.
DOI: 10.1039/D5CP00466G
314. Ya-Shan Huang, Hong-Lei Xu, Wen-Juan Tian, Zi-Sheng Li, Sílvia Escayola, Miquel Solà, Alvaro Muñoz-Castro, Zhong-Ming Sun. [Co3@Ge6Sn18]5−: A Giant σ-Aromatic Cluster Analogous to H3+ and Li3+. J. Am. Chem. Soc. 2025, 147, 11, 9407–9414.
DOI: 10.1021/jacs.4c16401
313. Mesías Orozco-Ic, P. L. Rodríguez-Kessler, A. Muñoz-Castro. Planar and Spherical Superatoms. Evaluating the aromaticity in Ag6Ru and Ag10Ru as electron precise clusters. ChemPhysChem, 2025, e202401118. DOI: 10.1002/cphc.202401118
312. Macarena Rojas-Poblete, Raúl Guajardo-Maturana, Alvaro Muñoz-Castro. Towards an efficient delayed fluorescence mechanism from charge transfer and local transitions. The role of heavy halide atoms. New J. Chem. 2025, 49, 5240-5247. DOI: 10.1039/D4NJ04975F
311. Alvaro Muñoz-Castro. Aromatic Trails. Persistence and Interplay Between Linked Spherical Aromatic Dicarboranes in Dimer to Hexamer Linear Arrays. Phys. Chem. Chem. Phys. 2025, 27, 5249-5255. DOI: 10.1039/D4CP03930K
310. Nicholas Charistos, Samuel R. Lawrence, Alvaro Muñoz-Castro. Global and Local Shielding/Deshielding Variation in C60 and C70 , Upon Reduction. Evaluation of 13C-NMR and Anisotropy Patterns. Chem. Eur. J. 2025, 31, e202404182. DOI: 10.1002/chem.202404182
309. P. L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Zn2-bicapped B7 wheel for the global minimum of B7Zn2 cluster. Phys. Chem. Chem. Phys. 2025, 27, 3583-3587.
DOI: 10.1039/D4CP04444D
308. Raul Guajardo Maturana, Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Insights into the Planarization of Benzo-Thianthrene rings. Electronic and Strain Effects and Resulting Aromatic Properties. J. Phys. Chem. A. 2025, 129, 1233-1239. DOI: 10.1021/acs.jpca.4c06257
307. Peter L. Rodriguez-Kessler, Alvaro Muñoz-Castro. [CuSn5Sb3]4- as a Dual Spherical-Spherical Aromatic Heterometallic Cluster Bridged via an Antiaromatic Cu2Sn2 Motif. Phys. Chem. Chem. Phys. 2025, 27, 2704-2710.
DOI: 10.1039/D4CP03591G
306. Renato Pereira Orenha, Alvaro Muñoz-Castro, Maurício Piotrowski, Giovanni Caramori, Renato Rocha, Renato Parreira. The Improved Skill of Rotaxanes to Recognize Cations: A Theoretical Perspective. ACS Phys. Chem Au 2025, 5, 183–194.
DOI: 10.1021/acsphyschemau.4c00090
305. Peter L. Rodriguez-Kessler, Alvaro Muñoz-Castro. [o-C2B10H10]3 as a Trimer Aggregate of Spherical Aromatic Carboranes. Persistence and Interplay Between Spherical Aromatic States. Chem. Phys. Lett. 2025, 858, 141752.
DOI: 10.1016/j.cplett.2024.141752
304. Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Nb11O15-. As a Persistent Spherical Antiaromatic Cluster. Chem. Phys. Lett. 2024, 857, 141687.
DOI: 10.1016/j.cplett.2024.141687
303. Alvaro Muñoz-Castro. C84Cl30, A Non-classical Fullerene Chloride Featuring Local Aromatic Motifs. Evaluation of 13C-NMR and Aromatic Patterns From DFT Calculations. Chem. Phys. Lett. 2024, 857, 141693. DOI: 10.1016/j.cplett.2024.141693
302. Yuvaraja Dibdalli, Mungalimane K. Amshumali, Elies Molins, Alvaro Muñoz-Castro, Cesar Morales-Verdejo. Synthesis, Reduction, and Hydrolysis of Zirconium Dimer and Trimer Complexes Derived from [(Cp*ZrCl2)2-as-Ic]: X-Ray crystal structure of [{(η⁵-C₅Me₅)Zr(μ₂-Cl)}₂-as-Ic] and [{(η⁵-C₅Me₅)Zr(μ₂-Cl)₂)}₃(μ-O)(µ-OH)]. Inorg. Chem. Comm. 2024, 170, 113331.
DOI: 10.1016/j.inoche.2024.113331
301. Wei-Xing Chen, Wen-Juan Tian, Zi-Sheng Li, John E. McGrady, Alvaro Muñoz-Castro, Zhong-Ming Sun. Capturing Aromatic Cyclo-Cr5 Plane in Large Main Group Metallic Cavities. Nature Synthesis. 2025, 4, 471-478. DOI: 10.1038/s44160-024-00711-5
300. Alvaro Muñoz-Castro. The Concentric Bond. Bonding Interaction in Concentric Structural Layers in Gold Superatoms. ChemPhysChem, e202400892.
299. Raul Guajardo-Maturana, Peter L. Rodríguez-Kessler, and Alvaro Muñoz-Castro. Nature of Cation-π Interaction in Mono- and Inverted Sandwich Ga(I) complexes. Insights into the Ga(I)-π Features and NMR Patterns from GaCp, GaCp*, [Ga2Cp]+, and [Ga2Cp*]+. Eur. J. Inorg. Chem., e202400400
298. Peter. L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Structure Search for Transition Metal Clusters. Towards a Rational Understanding of their Size-Dependent Properties. Inorg. Chim. Acta. 2025, 574, 122376.
DOI: 10.1016/j.ica.2024.122376
297. JianYu Wei, Peter L. Rodríguez-Kessler, Jean-Yves Saillard, and Alvaro Muñoz-Castro. Cuboctahedral Pd13 as a spherical Aromatic Noble Metal Core. Insights from ligand-protected [Pd13(Tr)6]2+ Cluster. Dalton. Trans. 2024,53, 16740-16746.
DOI: 10.1039/D4DT01633E
296. P.L. Rodríguez-Kessler, Salomon Rodríguez-Carrera, Alvaro Muñoz-Castro. Hydrogen Evolution Reaction on Ag12M icosahedrons: A DFT Study. Chem. Phys. Lett. 2024, 856, 141588.
DOI: 10.1016/j.cplett.2024.141588
295. Peter L. Rodríguez-Kessler, Jose Manuel Guevara-Vela, Tomas Rocha-Rinza, Mesías Orozco-Ic, David Olalde-Lopez, Alvaro Munoz-Castro. Ru-doped Agn (n= 1-13) clusters: Ag10Ru a 18-ve cluster superatom . Inorg. Chim. Acta, 2025, 574, 122349.
DOI: 10.1016/j.ica.2024.122349
294. Raul Guajardo Maturana, Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. On the Benzene-Ni10S20 Interaction. Host-guest Features and NMR Patterns Towards Prototypical Metal-Based Active Cavities. J. Phys. Chem. C 2024, 128, 15549–15557.
293. Jose Manuel Guevara-Vela, Miguel Gallegos, Tomas Rocha-Rinza, Alvaro Muñoz-Castro, Peter L. Rodríguez-Kessler, Angel Martín Pendas. New global minimum conformers for the Pt19 and Pt20 clusters.Considerations about the role of basis set quality and Hartree-Fock exchange.J. Mol. Model. 2024, 30, 310.
DOI: 10.1007/s00894-024-06099-5
292. Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro Intercluster B-H and B-B Aggregation in iso- and trans-[B20H18]2-. Spherical Aromaticity in Borane Dimers B20H18. Dalton. Transc. 2024, 53, 13960-13967.
DOI: 10.1039/D4DT01699H
291. Camila Muñoz Zuñiga, Vinicius C. Port, Carolina Olea Ulloa, Peter L. Rodríguez-Kessler, Renato L. T. Parreira, Giovanni F. Caramori, and Alvaro Muñoz-Castro. Anion Encapsulation with Micromolar Affinity on [Pd2L4]4+ Receptor Cage. Nature of Anion-Receptor Interaction and 1H-NMR Parameters from Dispersion-Corrected DFT Calculations. J. Phys. Chem. C, 2024, 128, 14017−14024.
290. Alvaro Muñoz-Castro. Beyond The Sphere. Au20(PR3)8 as a Spherical Aromatic Cuboctahedron Cluster Chem. Asian J. 2024, e202400670.
289. Salomon Rodıíguez-Carrera, P. L. Rodríguez-Kessler, F. Ambriz-Vargas, R. Garza-Hernández, R. Reséndiz-Ramírez, J. S. Martínez-Flores, A. Benitez-Lara, M. A. Martínez-Gameza, A. Muñoz-Castro. First principles study for Ag-based core-shell nanoclusters with 3d-5d transition metal cores for the oxygen reduction reaction. Inorg, Chim. Acta., 2024, 572, 122301.
DOI: 10.1016/j.ica.2024.122301
288. Macarena Gemita Rojas, Raúl Guajardo-Maturana, Luis Velásquez, Plinio Cantero-Lopez, Alvaro Muñoz-Castro. 4’–phenyl -2, 2’: 6’, 2’’-terpyridine Derivatives as Metal Chemosensors. Chelation and Fluorescence Capabilities Towards Zn(II), Cd(II), and Hg(II) From Experiment and Theory. J. Photochem. Photobiol., A, 2024, 457, 115885.
DOI: 10.1016/j.jphotochem.2024.115885
287. Mukundam Vanga, Alvaro Muñoz-Castro, H. V. Rasika Dias. Coinage metal complexes of a sterically encumbered anionic pyridylborate. Chem. Eur. J. 2024, 30, e202401204.
286. Raul Guajardo-Maturana, Desmond MacLeod Carey, Peter L. Rodríguez-Kessler, and Alvaro Muñoz-Castro. On the Variation of Cluster Core Characteristics by Endohedral Atom. Shape Variation in 8-ce [EAu4(PPh3)4]2+ (E=N, P, As, Sb) Clusters. Phys. Chem. Chem. Phys. 2024, 26, 20701-20708.
DOI: 10.1039/D4CP01465K
285. Raul Guajardo Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro. Which fits better?. Centered and non-Centered coordination modes of small endohedral Fullerenes, insights from Ti@C28 and Zr@C28. Inorg. Chim. Acta, 2024, 571, 122243.
DOI: 10.1016/j.ica.2024.122243
284. Rafael Lingas, Nickolas Charistos, Alvaro Muñoz-Castro. Charge Delocalization and Aromaticity of Doubly Charged Carbon Nanohoop Complexes with Fullerene C60 and Borospherene B40. Chem. Eur. J. 2024, 30, e202402027.
283. Raul Guajardo Maturana, Peter. L. Rodríguez-Kessler, and Alvaro Muñoz-Castro. On the Halide Aggregation into [Au4(PPh3)4]4+ Cluster Core. Insights from Structural, Optical and Interaction Energy Analysis in [(Ph3PAu)4X2]2+ and [(Ph3PAu)4X]3+ species (X=Cl-, Br-, I-)Au4X2. Phys. Chem. Chem. Phys. 2024, 26, 18828-18836.
DOI: 10.1039/D4CP01467G
282. [Editorial board special issue] Peter L. Rodríguez-Kessler and Alvaro Muñoz-Castro. Intermediate Intercluster Bond Orders. Electronic Communication in Au38(SR)24 Superatomic Molecules. ChemPhysChem. 2024, 25, e202400183.
281. David Olalde Lopez, P. L. Rodríguez-Kessler, Salomon Rodríıguez-Carrera, Alvaro Munoz-Castro. Hydrogen Storage Properties for Bimetallic Doped Boron Clusters M2B7 (M=Fe, Co, Ni). Int. J. Hydrogen Energy. 2024, in Press.
DOI: 10.1016/j.ijhydene.2024.05.429
280. Daniela Fonseca-Lopez, Johan D. Lozano, Mario A. Macías, Alvaro Muñoz-Castro, Desmond MacLeod-Carey, Edgar Nagles, John Hurtado. Biological Activity of Complexes Involving Nitro-Containing Ligands and Crystallographic-Theoretical Description of 3,5-DNB Complexes. Int. J. Mol. Sci. 2024, 25, 6536.
DOI: 10.3390/ijms25126536
279. Songlin Xue, Nikolay V. Tkachenko, Fan Wu, Xiaojuan Lv, Ningchao Liu, Alvaro Muñoz-Castro, So Ueno, Naoki Aratani, Zhen Shen, Hiroko Yamada, Alexander I Boldyrev, Fengxian Qiu. Conflicting Aromaticity in Crystalline Tri-Rhodium Organometallic Complex. Inorg. Chem. 2024, 63, 24, 11494-11500.
DOI: 10.1021/acs.inorgchem.4c01781
278. Alvaro Muñoz-Castro. [Ba4@Sn56]36- as a Main-Group Second-Order Superatom. Interpenetrated Dodecahedrons as a Three-Dimensional Cluster-of-Clusters Structure. Inorg. Chem. 2024, 63, 43, 20102–20107.
DOI: 10.1021/acs.inorgchem.4c00104
277. Agnieszka Varchmin, Alvaro Muñoz Castro, Ingo Ott. Gold(I) organometallics with N-heterocyclic carbene and alkyne ligands and their biological evaluation as prospective anticancer drugs. J. Organomet. Chem. 2024, 1012, 123148.
DOI: 10.1016/j.jorganchem.2024.123148
276. P. L. Rodríguez-Kessler, , Alejandro Vasquez-Espinal , A. R. Rodríguez-Domínguezc , J. L. Cabellos-Quiroz , Qi Yang,, Zi-Yu Li, Sheng-Gui Hee, A. Munoz-Castro. Structures of Ni doped Bn (n=1-12) clusters: A computational study. Inorg. Chim. Acta. 2024, 568, 122062.
DOI: 10.1016/j.ica.2024.122062
275. Cong-Cong Shu, Dariusz W. Szczepanik, Alvaro Muñoz-Castro, Miquel Solà, Zhong-Ming Sun. [K2(Bi@Pd12@Bi20)]4−: An Endohedral Inorganic Fullerene with Spherical Aromaticity. J. Am. Chem. Soc. 2024, 146, 20, 14166–14173.
DOI: 10.1021/jacs.4c03024
274. Francisca Claveria-Cadiz, Macarena Rojas-Poblete, Raúl Guajardo-Maturana, Alvaro Muñoz-Castro and Eduardo Schott. Electronic Pathway of the Exciplex Emitters. Unraveling the Emission properties from Theoretical Calculations. Journal of Photochemistry and Photobiology A: 2024, 452, 115547.
DOI: 10.1016/j.jphotochem.2024.115547
273. Peter L. Rodriguez-Kessler, Alvaro Muñoz-Castro. Ligand-Free Supermolecules. [Pd2@Ge18]4- and [Pd2@Sn18]4- as Multiple Bonded Zintl-Ions Cluster Based on Pd@Ge9 and Pd@Sn9 Assembled Units. Pd2Sn18. Nanoscale, 2024, 16, 5829-5835.
DOI: 10.1039/D4NR00220B
272. [Invited 25th anniversary PCCP] Peter L. Rodriguez-Kessler, Alvaro Muñoz-Castro. [Ag(Sn9–Sn9)]5- and [(η4-Sn9)Ag(η1-Sn9)]7-, as Aggregates of Spherical Aromatic Building Blocks. Persistence of Aromaticity Upon Cluster Gathering. Phys. Chem. Chem. Phys. 2024, 26, 8419-8425.
DOI: 10.1039/D3CP05394F





















































271. H. V. Rasika Dias, ORCID, Devaborniny Parasar, Andrey A. Yakovenko, Peter W. Stephens, Álvaro Muñoz-Castro, Mukundam Vanga, Pavel Mykhailiuk, and Evgeniy Slobodyanyuke. In situ studies of reversible solid-gas reactions of ethylene responsive silver pyrazolates. Chem. Sci., 2024.
DOI: 10.1039/D3SC04182D
270. Francisca Claveria-Cadiz, Raúl Guajardo-Maturana, Alvaro Muñoz-Castro. Unraveling the Delayed Fluorescent Mechanism of Copper(I) dyes from DFT Calculations. Results in Chemistry, 2023, 6, 101201.
DOI: 10.1016/j.rechem.2023.101201
269. Maria Laura Lucas Natal, Renato Pereira Orenha, Alvaro Muñoz-Castro, Giovanni Finoto Caramori, and Renato Luis Tame Parreira. Rational Design of Promising Candidates for Photoactive Layer in Polymer Solar Cells: Insights from Computation. J. Phys. Org. Chem., 2023, e4586.
DOI:10.1002/poc.4586
268 Amr A.A. Attia, Alexandru Lupan, R. Bruce King and Alvaro Muñoz-Castro. Dual Spherical Aromaticity in [M(η2-P4)2]+ (M=Cu, Ag, Au). Evaluation of Bonding Nature and Spherical Aromatic Character from Relativistic DFT Calculations. Inorg. Chem. Comm. 2024, 159, 111680.
DOI: 10.1016/j.inoche.2023.111680
267. Mukundam Vanga, Alvaro Muñoz-Castro. H. V. Rasika Dias. Classical gold carbonyl complexes in tetrahedral and trigonal-planar settings. Chem. Eur. J., 2023, e202303339.
266. Alvaro Muñoz-Castro. Second-Order Superatoms. Au52-PAP Featuring a Three-Dimensional Cluster-of-Clusters Core. Dalton. Trans., 2023,52, 17696-17700.
DOI: 10.1039/D3DT02693K
265. Yu-He Xu, Wen-Juan Tian, Alvaro Muñoz-Castro, Gernot Frenking, Zhong-Ming Sun. An all-metal fullerene: [K@Au12Sb20]5-. Science, 2023, 382, 6672, 840-843.
264. Mukundam Vanga, Vo Quang Huy Phan, Jiang Wu, Alvaro Muñoz-Castro, H. V. Rasika Dias. Thallium(I) complexes of tris(pyridyl)borates and a comparison to their pyrazolyl analogs. Inorg. Chem. 2023, 62, 45, 18563-18572.
DOI: 10.1021/acs.inorgchem.3c02805
263. Mulkundam Vanga, Benjamin T. Diroll, Alvaro Muñoz-Castro, H. V. Rasika Dias. Filling the gap with a bulky diaryl boron group: fluorinated and non-fluorinated copper pyrazolates fitted with a dimesityl boron moiety on the backbone. Dalton Trans. 2023, 52, 16356-16363.
DOI: 10.1039/D3DT03167E
262. José Manuel Guevara-Vela, Tomás Rocha-Rinza, Peter L. Rodríguez-Kessler, and Alvaro Muñoz-Castro. On the structure and electronic properties of Ptn clusters: new most stable structures for n=15-17. Phys. Chem. Chem. Phys. 2023, 25, 28835-28840.
DOI: 10.1039/d3cp04455f
261. Ricardo A. Murcia, Sandra M. Durán, Sandra M. Leal, Martha V. Roa, Jose D. Vargas, Laura V. Herrera, Álvaro Muñoz-Castro, Desmond MacLeod-Carey, John J. Hurtado. Co(II) and Cu(II) complexes containing 1,3-bis(benzotriazol-1-yl)-propan-2-ol as antifungal agent: Synthesis and biological activity. BMC Chemistry. 2023, 17, 135.
DOI: 10.1186/s13065-023-01037-7
260. [Book chapter] A Branch of Zintl Chemistry: Metal Clusters of Group 15 Elements Yu-He Xu, Nikolay V. Tkachenko, Alvaro Muñoz-Castro, Alexander I. Boldyrev, and Zhong-Ming Sun. Atomically Precise Nanochemistry, Editor(s):Rongchao Jin, De-en Jiang. Wiley., 2023, 395-422. DOI: 10.1002/9781119788676.ch13
259. Renato Pereira Orenha, Giovanni Finoto Caramori, Renato Luis Tame Parreira, and Alvaro Munoz–Castro. The Use of Molecular Electronic Structure Methods to Investigate Mechanically Interlocked Molecules. 2023, 25, 19409-19421.
DOI: 10.1039/D3CP02275G
258. Rafael Lingas, Nickolas Charistos, Alvaro Muñoz-Castro. Charge Delocalization and Aromaticity of Doubly Charged Double-walled Carbon Nanohoops. Phys. Chem. Chem. Phys. 2023, 25, 19481-19491.
DOI: 10.1039/D3CP01994B
257. Peter L. Rodríguez-Kessler, Alejandro Vázquez-Espinal, Alvaro Muñoz-Castro. Structure and stability of Cu-doped Boron clusters: Density Functional Theory Calculations Polyhedron. 2023, 243, 116538.
DOI: 10.1016/j.poly.2023.116538
256. Alexandre O. Ortolan, Leticia Madureira, Peter. L. Rodríguez-Kessler, Raul Guajardo Maturana, Carolina Olea Ulloa, Giovanni F. Caramori, Renato L. T. Parreira, and Alvaro Muñoz-Castro. The Nature of the Central Halide Encapsulation in Bambusuril Hosts (BU[6]). Structural and Interaction Energy Insights in BU[6].X- (X=Cl, Br, I) from Relativistic DFT Calculations. Inorg. Chim. Acta. 2023, 555, 121596.
DOI: 10.1016/j.ica.2023.121596
255. Alvaro Muñoz-Castro, and Ramiro Arratia-Perez. Spin-Orbit Effects in Cluster Chemistry. Considerations and Applications for Rationalization of their Properties. Chemical Physics Reviews, 2023, 4, 021313.
DOI: 10.1063/5.0145779
254. Peter L. Rodríguez-Kessler, Alejandro Vázquez-Espinal, Alvaro Muñoz-Castro. Structure and stability of Mo-doped Cun (n=1-12) clusters: DFT calculations. Inorg. Chim. Acta. 2023, 556, 121620.
DOI: 10.1016/j.ica.2023.121620
253. Desmond MacLeod-Carey, Peter L. Rodríguez-Kessler, and Alvaro Muñoz-Castro. Cl@Si20X20 cages. Evaluation of Encapsulation Nature, Structural Rigidity, and 29Si-NMR Patterns from Relativistic DFT Calculations. Phys. Chem. Chem. Phys. 2023,25, 19845-19852.
DOI: 10.1039/D3CP02371K
252. Amr A.A. Attia, Alvaro Muñoz-Castro, Alexandru Lupan, and R. Bruce King. Aromaticity in P8 Allotropes and (CH)8 Analogues: Significance of their 40 Valence Electrons?. Phys. Chem. Chem. Phys. 2023, 25, 9364-9372.
DOI: 10.1039/D3CP00147D
251.Stefan Knoppe, Alvaro Muñoz-Castro. Intermediate Silver Doping of Au25(SR)18. Variation of Electronic, Optical and Chiroptical Properties along Au25-xAgx(SH)18- (x=0-12) Stoichiometry from Relativistic DFT Calculations. Inorg. Chem. 2023,62, 18, 7079–7086.
DOI: 10.1021/acs.inorgchem.3c00485
250. Alvaro Muñoz-Castro, R. Bruce King. The Interplay of d and f Orbitals in the Chemistry of the Early Actinides: High Spin Versus Low Spin. Polyhedron. 2023, 553, 121527.
DOI: 10.1016/j.ica.2023.121527
249. Rafael Lingas, Nickolas Charistos, Alvaro Muñoz-Castro. Local and Global Aromaticity under Rotation. Analysis of Two- and Three-Dimensional Representative Carbon Nanostructures. Phys. Chem. Chem. Phys., 2023, 25, 14285-14293.
DOI: 10.1039/D3CP00569K
248. Renato Pereira Orenha, Saulo Samuel Pereira Furtado, Alvaro Muñoz–Castro, Maurício Jeomar Piotrowski, Giovanni Finoto Caramori, and Renato Luis Tame Parreira. Tuning Mechanically Interlocked Molecules to Recognize Anions and Cations: A Computational Study. Chem. Eur. J. 2023, e202203905.
247. Renato Pereira Orenha, Saulo Samuel Pereira Furtado, Alvaro Muñoz-Castro, Maurício Jeomar Piotrowski, Giovanni Finoto Caramori, Renato Luis Tame Parreira. Anion Recognition using Enhanced Halogen Bonding through Intramolecular Hydrogen Bonds – A Computational Insight. New J. Chem. 2023, 47, 4439-4447.
DOI: 10.1039/D2NJ05969J
246. Carolina Olea Ulloa, Alvaro Muñoz-Castro. Infinitene as Two Fused Helicoidal Trails of Fused rings. Evaluation of Magnetic behavior of [12]Infinitene and Anionic Species Displaying Global Aromaticity and Antiaromaticity. Phys. Chem. Chem. Phys. 2023, 25, 8190-8197.
DOI: 10.1039/D2CP06039F
245. Carolina Olea Ulloa, Raul Guajardo-Maturana, and Alvaro Muñoz-Castro. On the Cation- Capabilities of Infinitene (∞). Evaluation of Bonding and Circular Dichroism Properties for Infinitene-Ag(I)n (n=1-4) Complexes from Relativistic DFT Calculations. Polyhedron. 2023, 234, 116323.
DOI: 10.1016/j.poly.2023.116323
244. Jianyu Wei, Desmond MacLeod Carey, Jean-François Halet, Samia Kahlal, Jean-Yves Saillard, and Alvaro Muñoz-Castro. From 8- to 18-ce Superatoms. Evaluation of the Ligand-Protected W@Au12(dppe)6 Cluster Displaying a Distinctive Electronic and Optical Properties From Relativistic Calculations. Inorg. Chem. 2023, 62, 7, 3047–3055.
DOI: 10.1021/acs.inorgchem.2c03771
243. Carolina Olea Ulloa, Peter L. Rodríguez-Kessler, Raul Guajardo-Maturana, and Alvaro Muñoz-Castro. Nature of the Dative Nitrogen-Coinage Metal Bond in Molecular Motors. Evaluation of NHC-M⸱⸱⸱Pyrazine Bond (M=Cu, Ag, Au) from Relativistic DFT. Inorg. Chim. Acta. 2023, 549, 121401.
DOI: 10.1016/j.ica.2023.121401
242. [Book chapter] Nikolay Tkachenko, Zhong-Ming Sun, Alexander I Boldyrev, Alvaro Muñoz-Castro. Advances in cluster bonding: Bridging superatomic building blocks via intercluster bonds. In book: Atomic Clusters with Unusual Structure, Bonding and Reactivity. In Atomic Clusters with Unusual Structure, Bonding and Reactivity Theoretical Approaches, Computational Assessment and Applications, Editors: Pratim Chattaraj, Sudip Pan, Gabriel Merino.
ISBN: 9780128231012
241. Xu-Hui Yue, Wei-Xing Chen, Tao Yang, Alvaro Muñoz-Castro, Gernot Frenking, Zhong-Ming Sun. Carbon-free sandwich compounds based on arsenic and antimony with icosahedral metal cores. Nature Synthesis. 2023, 2, 423–429.
DOI: 10.1038/s44160-023-00247-0
240. Desmond Macleod-Carey, Alvaro Muñoz-Castro. Switch from Local and Global Aromatic Character in Möbius Carbon Nanobelts upon Dioxidation. Evaluation of Magnetic Behavior in Neutral and Charged species. Phys. Chem. Chem. Phys. 2023, 25, 4467-4471.
DOI: 10.1039/D2CP05326H
239. Peter L. Rodríguez-Kessler, Alvaro Muñoz-Castro, Adán R. Rodríguez-Domínguez, José Luis Cabellos. Structure effects of Pt15 clusters for the oxygen reduction reaction: First-principles calculations. Phys. Chem. Chem. Phys. 2023, 25, 4764-4772.
DOI: 10.1039/D2CP05188E
238. Brandon T. Watson, Mukundam Vanga, Anurag Noonikara-Poyil, Alvaro Muñoz-Castro, and H. V. Rasika Dias. Copper(I), silver(I), and gold(I) ethylene complexes of fluorinated and boron-methylated bis- and tris(pyridyl)borate chelators. Inorg. Chem. 2023, 62, 4, 1636–1648.
DOI: 10.1021/acs.inorgchem.2c04009
237. Ricardo A. Murcia, Desmond MacLeod-Carey, John Hurtado, Alvaro Muñoz-Castro. Formation of C60-SnI4 Adducts. Insights of the role of Halogen σ-hole and Tetrel-bonding in the Interaction Nature and from DFT calculations. Inorg. Chim. Acta. 2023, 545, 121277.
DOI: 10.1016/j.ica.2022.121277
236. Macarena Rojas-Poblete, Peter L. Rodríguez-Kessler, Raul Guajardo-Maturana, Carolina Olea Ulloa, and Alvaro Muñoz-Castro. Nature and Role of Formal Charge of the ion Inclusion in Hexanuclear Platinum(II) Host-Guest Species. Insights from Relativistic DFT Calculations. Inorg. Chim. Acta. 2023, 546, 121298.
DOI: 10.1016/j.ica.2022.121298
235.Demond Macleod-Carey, Alvaro Muñoz-Castro. Enabling Dual Aromaticity in Fused Nanobelts. Evaluation Magnetic behavior of Fused [10]CPP Units. Phys. Chem. Chem. Phys. 2022, 24, 26701-26707.
DOI: 10.1039/D2CP03667C
234. Tatiana Gomez, Alvaro Muñoz-Castro. Ligand-Dictated Cluster Core Characteristics in Au8Se2 Gold Selenido. Insights from Relativistic DFT. Inorg. Chim. Acta. 2022, 542, 121149.
DOI: 10.1016/j.ica.2022.121149
233. Alvaro Muñoz-Castro, H. V. Rasika Dias. Bonding and 13C-NMR properties of coinage metal tris(ethylene) and tris(norbornene) complexes. Evaluation of the role of relativistic effects from DFT calculations. J. Comp. Chem. 2022, 43, 1848-1855.
DOI: 10.1002/jcc.26987
232. Ya-Shan Huang, Ivan A. Popov, Alvaro Muñoz-Castro, Zhong-Ming Sun. [Nb@Ge13/14]3−: New Family Members of Ge-Based Intermetalloid Clusters. Chemistry Eur. J. 2022, 28, e202202192.
231. Peter, Alvaro Muñoz-Castro. Structural and electronic properties for Be-doped Ptn (n = 1−12) clusters obtained by DFT calculations. Phys. Chem. Chem. Phys. 2022, 24, 7856-7861.
DOI: 10.1039/D1CP05410D
230. Alvaro Muñoz-Castro. N-Heterocyclic Carbene Derivatives to Modify Gold Superatoms Characteristics. Tailorable Electronic and Optical Properties for [Au11(PPh3)7LCl2]+ as Cluster from Relativistic DFT. Phys. Chem. Chem. Phys. 2022, 24, 5965-5973.
DOI: 10.1039/D1CP04310B
229. Wei-Xing Chen, Nikolay V. Tkachenko, Alvaro Muñoz-Castro, Alexander I. Boldyrev, and Zhong-Ming Sun. Ruthenium-mediated assembly and enhanced stability of heterometallic polystannides [Ru2Sn19]4- and [Ru2Sn20]6-. NanoResearch. 2022, 15, 5705-5711.
DOI: 10.1007/s12274-022-4178-9
228. Hong-Lei Xu, Nikolay V. Tkachenko, Alvaro Muñoz-Castro, Alexander I. Boldyrev, Zhong-Ming Sun.. Symmetry collapse due to the presence of multiple local aromaticity in Ge244-. Nat. Chem. 2022, 13, 2149.
DOI: 10.1038/s41467-022-29626-5
227. Mesías Orozco-Ic, Jorge Barroso, Nickolas D. Charistos, Rafael Islas, Alvaro Muñoz-Castro, Dage Sundholm, Gabriel Merino. Core-electron contributions to the molecular magnetic response. Phys. Chem. Chem. Phys. 2022, 24, 12158-12166.
DOI: 10.1039/D1CP05713H
226. Wei-Xing Chen, Nikolay Tkachenko, Alvaro Muñoz-Castro; Alexander Boldyrev, Zhong-Ming Sun. Sn368‒: A 2.7 nm Naked Tin Rod. Chem. Comm. 2022, 58, 6223-6226.
DOI: 10.1039/D2CC01745H
225. Alvaro Muñoz-Castro. Ligand-Core Interaction in Ligand-Protected Ag25(XR)18 (X= S, Se, Te) Superatoms. Evaluation of Anchor Atom Role via Relativistic DFT Calculations. Phys. Chem. Chem. Phys. 2022, 24, 17233-17241.
DOI: 10.1039/D2CP01058E
224. Jianyu Wei, Samia Kahlal, Jean-François Halet, Alvaro Muñoz-Castro, Jean-Yves Saillard. Ligand-induced cuboctahedral vs. icosahedral core isomerism within 8-electron NHC-protected gold nanoclusters. Inorg. Chem. 2022, 61, 23, 8623-8628.
DOI: 10.1021/acs.inorgchem.2c01022
223. Renato Parreira, Giovanni Caramori, Leticia Madureira, Raul Guajardo-Maturana and Alvaro Muñoz-Castro. Analysis of the Host-Guest Complex Formation Involving the HexaCage6+ Cavity. Insights from Dispersion Corrected DFT Calculations. J Nanostructure Chem. 2022, 12, 1143-1154.
DOI: 10.1007/s40097-022-00497-y
222. Jianyu Wei, Jean-François Halet, Samia Kahlal, Alvaro Muñoz-Castro and Jean-Yves Saillard. Insight into The Stability, Electronic and Optical Properties of N-Heterocyclic Carbene Derivatives from Halogen/Phosphine Protected Au13 Superatomic. J. Phys. Chem. A.. 2022, 126, 536-545. DOI: 10.1021/acs.jpca.1c09084
221. Anurag Noonikara Poyil, Alvaro Muñoz-Castro, H. V. Rasika Dias. Acetylene, terminal and internal alkyne complexes and azide-alkyne cycloaddition chemistry of copper(I) supported by a bis(pyrazolyl)borate Molecules 2022, 27, 16.
DOI: 10.3390/molecules27010016
220. Mukundam Vanga, Alvaro Muñoz-Castro and H. V. Rasika Dias. Fluorinated tris(pyridyl)borate ligand support on coinage metals. Dalton Trans. 2022, accepted.
DOI: 10.1039/D1DT04136C
219. Raul Guajardo Maturana, Alexandre O. Ortolan, Peter. L. Rodríguez-Kessler, Giovanni F. Caramori, Renato L. T. Parreira and Alvaro Muñoz-Castro. Nature of Hydride and Halide Encapsulation in Ag8 Cages. Insights from Structural and Interaction Energy [Ag8(X){S2P(OiPr)2}6]+ (X=H-, F-, Cl-, Br-, I-) from Relativistic DFT Calculations. Phys. Chem. Chem. Phys., 2022, 24, 452-458.
DOI: 10.1039/D1CP04249A
218. Alvaro Muñoz-Castro. On the Ligand Role in Determining the Compact or Extended Metallic Core Architecture in Gold Superatoms. Evaluation of Electronic and Optical Properties from Relativistic DFT for [Au11(dppp)5]3+ and [Au11(dppe)6]3+ Clusters. Polyhedron, 2022, 211, 115572.
DOI: 10.1016/j.poly.2021.115572
217. [Book Chapter] Giovanni F. Caramori, Alvaro Muñoz-Castro. Box-Shaped Hosts: Evaluation of the Interaction Nature and Host Characteristics of ExBox Derivatives in Host-Guest Complexes from Computational Methods. In Chemical Reactivity in Confined Systems: Theory, Modelling and Applications, Pratim Kumar Chattaraj, Debdutta Chakraborty (Eds). Wiley, 2021. DOI: 10.1002/9781119683353.ch20
216. Nikolay V. Tkachenko, Alvaro Muñoz-Castro and Alexander I. Boldyrev. Occurrence of Double Bond in π-Aromatic Rings: An Easy Way to Design Doubly Aromatic Carbon-Metal Structures. Molecules 2021, 26, 7232;
DOI: 10.3390/molecules26237232
215. Peter L. Rodríguez-Kessler, Adán R. Rodríguez-Domínguez, and Alvaro Muñoz-Castro. Structural Evolution and Electronic Properties of Intermediate Sized Tin (𝑛=33-60) Clusters. Adv. Theory Simul. 2021, 4, 2100283.
214. H. V. Rasika Dias; Anurag Noonikara-Poyil; Alvaro Muñoz-Castro; Andrii Boretskyi; Pavel Mykhailiuk. When SF5 outplays CF3: Effects of pentafluorosulfanyl decorated scorpionates on copper. Chem. Sci. 2021,12, 14618-14623
DOI: 10.1039/D1SC04846E
213. [Invited Prof. Boldyrev special issue] Nikita Fedik, D.V. Steglenko, A. Muñoz-Castro, R.M. Minyaev, V.I. Minkin. Band Gap Engineering and 14 Electron Superatoms in 2D Superoctahedral Boranes B4X2 (B, N, P, As, Sb). J. Chem. Phys. C. 2021, 125, 31, 17280–17290
212. Ai-Min Li, Yi Wang, Peter Zavalij, Yu-Sheng Chen, Alvaro Muñoz-Castro, and Bryan W. Eichhorn, Contrasting Bonding Extremes in Two [Ge6]n- Complexes: A Wadian polyhedron (n = 2) versus a Hydrocarbon-like 2c−2e Polygermanide (n = 12). Inorg. Chem. 2021, 60, 14679-14705 DOI: 10.1021/acs.inorgchem.1c01799
211. Alvaro Muñoz-Castro. Au70S20(PPh3)12 as Superatomic Analog to 18-electron Transition-Metal Complexes. Z. anorg. allg. Chem. 2021, 647, 1819-1823
210. Vanessa Molina, Juan Luis Arroyo, Desmond MacLeod-Carey, Alvaro Muñoz-Castro, Cesar Morales-Verdejo. Catalytic effect of [Cu6(dmPz)6(OH)6] on the thermal decomposition of ammonium perchlorate. Polyhedron, 2021, 209, 115464.
DOI: 10.1016/j.poly.2021.115464
209. [Invited] Desmond MacLeod Carey, Alvaro Muñoz-Castro. On the Aromaticity and 13C-NMR pattern of Pentagonal-Pyramidal Hexamethylbenzene Dication C6(CH3)62+. A C5(CH3)5- - CCH33+ Aggregate. Chemistry 2021, 3, 1363-1370. DOI: 10.3390/chemistry3040097
208. Nikolay V. Tkachenko, Maksim Kulichenko, Ivan A. Popov, Alvaro Muñoz-Castro, Zhong-Ming Sun and Alexander I. Boldyrev Bridging Aromatic/Antiaromatic Units. Recent Advances in Aromaticity and Antiaromaticity in Main-group and Transition-metal clusters. Eur. J. Inorg. Chem. 2021, 2021, 4239-4250
207. Macarena Rojas-Poblete, P. L. Rodríguez-Kessler, Raul Guajardo-Maturana and Alvaro Muñoz-Castro. Coinage-Metal Pillarplexes Hosts. Insights into Host-guest interaction Nature and Luminescence Quenching Effects. Phys. Chem. Chem. Phys. 2021, 23, 15917-15924.
DOI: 10.1039/D1CP00849H
206. P. L. Rodríguez-Kessler, Macarena Rojas-Poblete, Alvaro Muñoz-Castro. Evaluation of Ultrasmall Coinage Metal M13(dppe)6 M= Cu, Ag, Au Clusters. Bonding, Structural and Optical Properties from Relativistic DFT Calculations. Phys. Chem. Chem. Phys. 2021, 23, 18035-18043. DOI: 10.1039/D1CP02451E
205. P. L. Rodríguez-Kessler, Adán R. Rodríguez-Domínguez, Desmond MacLeod-Carey, Alvaro Muñoz-Castro. Reply to ‘Comment on ‘‘Structural characterization, reactivity, and vibrational properties of silver clusters: A new global minimum for Ag16’’’ by P. V. Nhat, N. T. Si, L. V. Duong and M. T. Nguyen, Phys. Chem. Chem. Phys., 2021, 23, DOI: D1CP00646K. Phys. Chem. Chem. Phys. 2021, 23, 12904-12906.
DOI: 10.1039/d1cp01481a
204. Yu-He Xu, Nikolay V. Tkachenko, Lei Qiao, Alvaro Muñoz-Castro, Alexander I. Boldyrev, and Zhong-Ming Sun. Ternary Aromatic and Anti-aromatic Clusters Stemmed from a hypho-Zintl Precursor [Sn2Sb5]3-. Nature Comm. 2021, 12, 4465
DOI: 10.1038/s41467-021-24706-4
203. Alvaro Muñoz-Castro. Aromaticity in Phenyl Decorated closo-Monocarboranes. Planar-Spherical Aggregates Involving seven to twelve–vertex Cage. J. Phys. Chem. A. 2021, 125, 4861-4866.
202. Jianyu Wei, Peter Rodríguez-Kessler, Jean-François Halet, Samia Kahlal, Alvaro Muñoz-Castro and Jean-Yves Saillard On Heteronuclear Isoelectronic Alternatives to Au13(dppe)5Cl2: Electronic and Optical Properties of the 18-electron Os@Au12(dppe)5Cl2 Cluster from Relativistic DFT Computations. Inorg. Chem. 2021, 2021, 60, 11, 8173–8180.
DOI: 10.1021/acs.inorgchem.1c00799
201. [Book Chapter] Maksim Kulichenko, Nikita Fedik, Nikolay V. Tkachenko, Alvaro Muñoz-Castro, Zhong-Ming Sun, Alexander I. Boldyrev. Spherical aromaticity in inorganic chemistry. In AROMATICITY MODERN COMPUTATIONAL METHODS AND APPLICATIONS, Israel Fernandez Ed. Elsevier, 2021.
ISBN: 9780128227435
200. Alvaro Muñoz-Castro, Desmond MacLeod-Carey, Ramiro Arratia-Perez, Relativistic Effects on Dative Carbon-Coinage Metal Bond. Evaluation of NHC-MCl (M=Cu, Ag, Au) from Relativistic DFT. Polyhedron, 2020, 197 (2021), 115020.
DOI: 10.1016/j.poly.2021.115020
199. Sílvia Escayola, Albert Poater Alvaro Muñoz-Castro and Miquel Solà. An Unprecedented π-Electronic Circuit Involving an Odd Number of Carbon Atoms in a Grossly Warped Non-Planar Nanographene. Chem. Commun., 2021,57, 3087-3090.
DOI: 10.1039/D1CC00593F
198. Peter Rodríguez-Kessler, Adán Rodríguez-Domínguez, Alvaro Muñoz-Castro. Exploring the Size-Dependent Hydrogen Storage Property on Ti-Doped Bn Clusters. Temperature Controlled H2 Release. Adv. Theory Simul. 2021, 4, 2100043.
197. Hong-Lei Xu, Nikolay V. Tkachenko, Alvaro Muñoz-Castro, Alexander I. Boldyrev, and Zhong-Ming Sun. [Sn8]6--bridged mixed-valence Zn(I)/Zn(II) in {[K2ZnSn8(ZnMes)]24- Inverse Sandwich-Type Cluster Supported by ZnI-ZnI Bond. Angew. Chem. Int. Ed. 2021, 60, 1 – 7.
196. Peter L. Rodríguez-Kessler, Adán R. Rodríguez-Domínguez, and Alvaro Muñoz-Castro, Systematic Cluster Growth: A Structure Search Method for Transition Metal Clusters. Phys. Chem. Chem. Phys., 2021,23, 4935-4943.
DOI: 10.1039/D0CP06179D
195. Cong-cong Shu, Lei Qiao, Alvaro Muñoz-Castro, Zhong-Ming Sun. [As3M(As3Pb3)]3- (M= Nb, Ta): Ternary Heterometallic Clusters with Electron-Deficient Transition Atoms and Aromatic [Pb3]2-. Chin. J. Chem. 2021, 39, 1953—1957.
194. Lei Qiao, Dandan Chen, Jun Zhu, Alvaro Muñoz-Castro, Zhong-Ming Sun. [Bi6Mo3(CO)9]4−: A multiple local σ-aromatic cluster containing distorted Bi6 triangular prism. Chem. Commun., 2021, Chem. Commun., 2021,57, 3656-3659.
DOI: 10.1039/D1CC00734C
193. Peter L. Rodríguez-Kessler, Adán R. Rodríguez-Domínguez, and Alvaro Muñoz-Castro. On the Structure and Reactivity of PtnCun (n = 1-7) Alloy. Phys. Chem. Chem. Phys., 2021,23, 7233-7239.
DOI: 10.1039/D1CP00379H
192. Rafael Lingas, Nickolas D. Charistos and Alvaro Muñoz-Castro. Aromaticity in ortho and meta 8-cycloparaphenylene dications: An induced magnetic field analysis with localized and delocalized orbitals. Chem.Phys. Chem. 2021, 22, 1–12.
191. Jiang Wu, Anurag Noonikara-Poyil, Alvaro Muñoz-Castro, H. V. Rasika Dias, Gold(I) Ethylene complexes supported by electron rich scorpionates. Chem. Commun., 2021,57, 978-981.
DOI: 10.1039/D0CC07717H
190. Ahmed H. Elashkar, Devaborniny Parasar, Alvaro Muñoz-Castro, Cara M. Doherty, Matthew G. Cowan, H. V. Rasika Dias, Isolable 1-butene copper(I) complexes and 1-butene/butane separation using structurally adaptable copper pyrazolates. ChemPlusChem. 2021, 86, 364-372.
189. Dandan Chen, Dariusz W. Szczepanik, Jun Zhu, Alvaro Muñoz-Castro, Miquel Sola Aromaticity Survival in Hydrofullerenes: The Case of C66H4 with its π-Aromatic Circuits. Chem. Eur. J. 2020, 27, 802-808.
188. Cristina A. Barboza, Adrian Gambetta, Ramiro Arratia-Perez, Peter L. Rodriguez-Kessler, Alvaro Muñoz-Castro, Desmond MacLeod-Carey. Structural, Electronic and Magnetic Properties of Copper(I) Cubic Clusters. Polyhedron, 2021, 195, 114878.
DOI: 10.1016/j.poly.2020.114878.
187. Renato P. Orenha, Glaucio R. Nagurniak, Matheus C. Colaço, Giovanni F. Caramori, Maurício J. Piotrowski, Aluno Mauricio, Alvaro Muñoz–Castro, Breno de Almeida Silva, Benjamim José Esteves, and Renato L. T. Parreira. The Simultaneous Recognition Mechanism of Cations and Anions from Macrocyclic–Iodine Structures: Insights from Dispersion corrected–DFT Calculations. Phys. Chem. Chem. Phys. 2021, 195, 114878.
DOI: 10.1039/D0CP04291A
186. Alexandre Osmar Ortolan, Giovanni Finoto Caramori, Renato Luis Tame Parreira, Renato Pereira Orenha, Alvaro Muñoz-Castro and Gernot Frenking. Bonding Situation In Heteromultimetallic Carbonyl Complexes. Dalton. Trans., 2020, 49, 16762-16771.
DOI: 10.1039/D0DT02916E
185. Giovanni F. Caramori, Ina Østrø, Alexandre O. Ortolan, Glaúcio R. Nagurniak, Vitor M. Besen, Alvaro Muñoz-Castro, Renato P. Orenha, Renato L. T. Parreira, and Sérgio E. Galembec. The Usefulness of Energy Decomposition Schemes to rationalize Host-Guest Interactions. Dalton. Trans., 2020, 49, 17457-17471.
DOI: 10.1039/D0DT03518A
184. Alvaro Muñoz-Castro, Guocang Wang, Tharun Teja Ponduru, H. V. Rasika Dias. Synthesis and Characterization of N-Heterocyclic Carbene-M∙∙∙OEt2 Complexes (M = Cu, Ag, Au). Analysis of Solvated Auxiliary-Ligand Free [(NHC)M]+ Species. Phys. Chem. Chem. Phys., 2021, 23, 1577-1583.
DOI: 10.1039/D0CP05222A
183. P. L. Rodríguez-Kessler, A. R. Rodríguez-Domínguez, Desmond MacLeod Carey, Alvaro Muñoz-Castro. Structural characterization, reactivity, and vibrational properties of silver clusters: A new global minimum for Ag16. Phys. Chem. Chem. Phys., 2020, 22, 27255-27262.
DOI: 10.1039/D0CP04018E
182. Qi Wang, Jean-François Halet, Samia Kahlal, Alvaro Muñoz-Castro and Jean-Yves Saillard. Electron count and electronic structure of bare icosahedral Au32 and Au33 ionic nanoclusters and ligated derivatives. Stable models with intermediate superatomic shell fillings. Phys. Chem. Chem. Phys. 2020, 22, 20751-20757.
DOI: 10.1039/D0CP03735D
181. Jianyu Wei, Jean-François Halet, Samia Kahlal, Jean-Yves Saillard and Alvaro Muñoz-Castro. Towards the Formation of N-Heterocyclic Carbene-Protected Gold Clusters of Various Nuclearities. A Comparison with their Phosphine-Protected Analogs from DFT Calculations. Inorg. Chem., 2020, 59, 20, 15240–15249.
DOI: 10.1021/acs.inorgchem.0c02219
180. Hong-Lei Xu, Ivan A. Popov, Nikolay V. Tkachenko, Zi-Chuan Wang, Alvaro Muñoz-Castro, Alexander I. Boldyrev, and Zhong-Ming Sun. σ-Aromaticity-Induced Stabilization of Heterometallic Supertetrahedral Clusters [Zn6Ge16]4– and [Cd6Ge16]4–. Angew. Chem. Int. Ed. 2020, 59, 17286-17290.
179. On the 13C-NMR Chemical Shift Anisotropy (CSA) Patterns and Aromatic Character in Strained Fullerenes. Computational Analysis of D6h/D2d-C36 Fullerene. Int. J. Quant. Chem. 2021, 121, e26437.
DOI: 10.1002/qua.26437
178. Maksim Kulichenko, Nikita Fedik, Anna Monfredini, Alvaro Muñoz-Castro, Davide Balestri, Alexander I. Boldyrev, Giovanni Maestri. “Bottled” doubly aromatic trinuclear [Pd2Ru]+ complexes. Chem. Sci., 2021, 2021,12, 477-486.
DOI: 10.1039/D0SC04469E
177. Ai-Min Li, Yi Wang, Peter Zavalij, Fu Chen, Alvaro Muñoz-Castro, and Bryan W. Eichhorn. [Cp*RuPb11]3– and [Cu@Cp*RuPb11]2–: Centered and Non-Centered Transition-Metal Substituted Zintl Icosahedra. Chem. Comm., 2020, 56, 10859-10862.
DOI: 10.1039/D0CC03656K
176. P. L. Rodríguez-Kessler, A. R. Rodríguez-Domínguez, J. A. Morato-Márquez, Filiberto Ortiz-Chi, Desmond MacLeod Carey, Alvaro Muñoz-Castro. On the Search of Small Cu-Ru Atomically Precise Superatoms. Cu10Ru Cluster as a Stable 18-ve Endohedral Structure Chem. Phys. Lett. 2020, 754, 137721.
DOI: 10.1016/j.cplett.2020.137721
175. Alvaro Muñoz-Castro and R. Bruce King. Th@C86, Th@C82, Th@C80, and Th@C76. Role of Thorium Encapsulation in Determining Spherical Aromatic and Bonding Properties on Medium-Sized Endohedral Metallic Fullerenes. Phys. Chem. Chem. Phys. 2020, 22, 23920-23928.
DOI: 10.1039/D0CP03784B
174. Hong-Lei Xu, Nikolay V. Tkachenko, Zi-Chuan Wang, Wei-Xing Chen, Lei Qiao, Alvaro Muñoz-Castro, Alexander I. Boldyrev & Zhong-Ming Sun. A Sandwich Type Cluster Containing Ge©Pd3 Interlayer Flanked by Aromatic Nonagermanide Caps. Nat. Comm., 2020, 11, 5286.
DOI: 10.1038/s41467-020-19079-z
173. Ina Ostrom, Alexandre O. Ortolan, Giovanni F. Caramori, Mark Mascal, Alvaro Muñoz-Castro and Renato L. T. Parreira. In Silico Design of Cylindrophanes: The Role of Functional Groups in a Fluoride Selective Host. ChemPhysChem., 2020,21 1989-2005.
172. José Aminadat Morato-Márquez; Srinivas Godavarthi; Claudia G. Espinoza-González; J. Gilberto Torres-Torres; A. R. Rodríguez-Domínguez; Alvaro Muñoz-Castro; Peter L. Rodríguez-Kessler. Structural Characterization and Electronic Properties of Ru-Doped Cun (n = 1−12) Clusters. Chem. Phys. Lett. 2020, 754, 137677.
DOI: 10.1016/j.cplett.2020.137677
171. G Naaresh Reddy, Rakesh Parida, Alvaro Muñoz-Castro and Santanab Giri. Doped Deltahedral Organo-Zintl Superalkali Cations., 2020, 759, 137952.
DOI: 10.1016/j.cplett.2020.137952
170. Peter L. Rodríguez-Kessler, Nickolas D. Charistos, R. Bruce King, Alvaro Muñoz-Castro, On the Formation of Spherical Aromatic Endohedral Buckminsterfullerene. Evaluation of M@C60 (M=Cr, Mo, W) From Relativistic DFT Calculations. Phys. Chem. Chem. Phys. 2020, 22, 14268-14275.
DOI: 10.1039/D0CP02475A
169. Éder H. da Silva, Renato P. Orenha, Alvaro Muñoz‐Castro, Giovanni Finoto Caramori, Matheus Cachoeira Colaço, and Renato L. T. Parreira. Theoretical Study of Chloride Complexes with Hybrid Macrocycles. New J. Chem., 2021, 45, 463-470.
DOI: 10.1039/D0NJ05234E
168. Glaucio Régis Nagurniak, Maurício Jeomar Piotrowski, Alvaro Muñoz-Castro, Renato Luis Tame Parreira, João Baptista S. Cascaldi and Giovanni Finoto Caramori. What is the driving force behind molecular triangles and its guests? A quantum chemical perspective at Host-Guest interactions. Phys. Chem. Chem. Phys., 2020, 22, 19213-19222.
DOI: 10.1039/D0CP01821J
167. Desmond MacLeod Carey, Peter L. Rodríguez‐Kessler, Alvaro Muñoz-Castro. Visualizing NMR-shielding Effect in Fullerene-ZnPc Aggregates. Characteristic Patterns of ZnP-Based Hosts and Encapsulation Nature From DFT Calculations. . Int. J. Quant. Chem., 2021, 121, e26500.
DOI: 10.1002/qua.26500
166. Nickolas Charistos, Alvaro Muñoz-Castro. Induced Magnetic Field in sp-Hybridized Carbon Rings: Analysis of Double Aromaticity and Antiaromaticity in Cyclo[2n]carbon Allotropes. Phys. Chem. Chem. Phys., 2020, 22, 9240-9249.
DOI: 10.1039/D0CP01252A
165. [Cover] Alvaro Muñoz-Castro. Interplay Between Planar and Spherical Aromaticity. Shielding Cone Behavior in Dual Planar-Planar, Planar-Spherical and Spherical-Spherical Aromatics. Chem. Phys. Chem., 2020, 21, 1384-1387.
164. Desmond MacLeod-Carey, Alvaro Muñoz-Castro. Evaluation of C60 and C70 Analogs Bearing Cyclo‐meta‐Phenylene Faces as Improved devices for Polymer: Fullerene solar cells from DFT Calculations. Dyes and Pigments., 2021, 184, 108782.
DOI: 10.1016/j.dyepig.2020.108782
163. Peter Rodríguez-Kessler, Pedro Alonso-Dávila, Jorge Barroso, Alvaro Muñoz-Castro, Faustino Aguilera-Granja, Adán Rodríguez-Domínguez, Structural, Electronic and Catalytic Properties of Bimetallic PtnAgn (n = 1-7) Clusters" J. Alloys Compd., 2020, 845, 155897.
DOI: 10.1016/j.jallcom.2020.155897
162. Rakesh Parida; G Naaresh Reddy; Edison Osorio; Alvaro Muñoz-Castro; Sukanta Mondal, Santanab Giri. Unique Magnetic Shielding and Bonding in Pnicogen Nortricyclane Zintl Clusters. Chem. Phys. Lett., 2020, 749, 137414.
DOI: 10.1016/j.cplett.2020.137414
161. Juan F. Torres, Nestor J. Bello-Vieda, Mario A. Macias, Renato Rabelo, Francisco Lloret, Alvaro Muñoz-Castro and John Hurtado. Intermolecular Interaction Energies and Magnetic Properties of Spin-isolated Multinuclear Cu(II) Complexes. Acta Cryst. B., 2020, B76, 166-176.
DOI: 10.1107/S2052520620001225
160. Z.-H. Wang, N. Tkachenko, L. Qiao, E. Matito, Alvaro Muñoz-Castro, A. Boldyrev, Z. Sun, All-Metal σ-Antiaromaticity in Dimeric Cluster Anion {[CuGe9(Mes)]2}4-. Chem. Commun., 2020, 56, 6583-6586.
DOI: 10.1039/D0CC02525A
159. Nikolay V. Tkachenko, Xiang-Wen Zhang, Lei Qiao, Cong-Cong Shu, Alvaro Muñoz-Castro, Zhong-Ming Sun, Alexander I. Boldyrev.. Spherical aromaticity of all-metal [Bi@In8Bi12]3-/5- clusters. Chem. Eur. J.,2020, 26, 2073 –2079.
158. Ai-Min Li, Ph.D; Yi Wang; Domonique O Downing; Fu Chen; Peter Y Zavalij, Alvaro Muñoz-Castro, Bryan W Eichhorn. Endohedral Plumbaspherenes of the Group 9 Metals: Synthesis, Structure and Properties of the [M@Pb12]3- (M = Co, Rh, Ir) Ions. Chem. Eur. J., 2020, 26, 5824 –5833
157. Maksim Kulichenko, Nikita Fedik, Alexander Boldyrev, Alvaro Muñoz-Castro. Expansion of aromaticity magnetic criteria on multi-layer structures. Magnetic response and spherical aromaticity of Matryoshka-like [Sn@Cu12@Sn20]12- cluster. Chem. Eur. J., 2020, 26, 2263 –2268.
156. Alvaro Muñoz-Castro. Triple 1D≡1D Superatomic Bonding. Au22(dppp)6 as a Π4- and Δ2-Triply Bonded Cluster Based on Au11 Assembled Units. Phys. Chem. Chem. Phys., 2020, 22, 1422-1426.
DOI: 10.1039/C9CP05790K
155. Renato Pereira Orenha, Nelson Henrique Morgon, Julia Contreras–García, Graziele Cappato Guerra Silva, Glaucio Régis Nagurniak, Maurício Jeomar Piotrowski, Giovanni Finotto Caramori, Alvaro Muñoz-Castro and Renato Luis Tame Parreira. How the Acidic Milieu Interferes in the Capability of Ruthenium Nitrosyl Complexes to Release Nitric Oxide. New J. Chem., 2020, 44, 773-779.
DOI: 10.1039/C9NJ04643G
154. Chandrakanta Dash, Guocang Wang, Alvaro Muñoz-Castro, Tharun T. Ponduru, Adway O. Zacharias, Muhammed Yousufuddin, H. V. Rasika Dias. Organic azide, and auxiliary ligand free complexes of coinage metals supported by N-heterocyclic carbenes. Inorg. Chem. 2020, 59, 4, 2188–2199.
DOI: 10.1021/acs.inorgchem.9b02771
153. Nikita Fedik, Alexander Boldyrev and Alvaro Muñoz-Castro. Aromatic Character of [Au13]5+ and [MAu12]4+/6+ (M=Pd,Pt) Core in Ligand Protected Gold Nanoclusters. Interplay Between Spherical and Planar σ-Aromatics. Phys. Chem. Chem. Phys. 2019, 21, 25215-25219.
DOI: 10.1039/C9CP04477A
152: Johanna Camacho Gonzalez, Sukanta Mondal, Fernanda Ocayo, Raul Guajardo Maturana, and Alvaro Muñoz-Castro. Nature of C60 and C70 Fullerene Encapsulation in a Porphyrin-and Metalloporphyrin-Based Cage. Insights from Dispersion Corrected DFT Calculations. Int J Quantum Chem., 2020,120, e26080.
DOI: 10.1002/qua.26080
151:Cristian Linares-Flores, Macarena Rojas, Ramiro Arratia-Perez, Alvaro Muñoz-Castro and Raul Guajardo Maturana. Role of Donor-Acceptor Functional Groups in N3P3 Cyclic-Triphosphazene Backbone. Unraveling Bonding Characteristics from Natural Orbitals Within an ETS-NOCV Scheme. Int. J. Quantum Chem., 2020,120, e26057.
DOI: 10.1002/qua.26057
150: Mesías Orozco-Ic, Jorge Barroso, Nickolas D. Charistos, Alvaro Muñoz-Castro, and Gabriel Merino. Consequences of the curvature on the induced magnetic field: Case of helicenes. Chem. Eur. J., 2020. 26, 326-330.
149: Constanza Gonzalez Quintana, Fernanda Ocayo, Raul Guajardo Maturana, John J. Hurtado, Alvaro Muñoz-Castro. "Nature of Mercury Inclusion in Intermediate 6-ve [M@Au8Hgx(PPh3)8]q (M = Au, Pd, Pt; x= 0-2) Protected Gold Superatoms. Insights from Relativistic DFT Calculations".
Int. J. Quantum Chem., 2020, 120, e26068.
DOI: 10.1002/qua.26068
148: Desmond MacLeod Carey, Alvaro Muñoz-Castro. "Evaluation of N-Heterocyclic Carbene Counterparts of Classical Gold Clusters; Bonding Properties of Octahedral CAu6, Icosahedral Au13Cl2, and Bi-icosahedral Au25Cl2 Cores from Relativistic DFT Calculations".
J. Phys. chem. C, 2019, 123, 12466-12473.
147: Nelson Nuñez-Dallos, Alvaro Muñoz-Castro, Mauricio Fuentealba, Edwin G. Perez, John J. Hurtado. "Facile synthesis of a luminescent copper(I) coordination polymer containing a flexible benzotriazole-based ligand: an effective catalyst for three-component azide-alkyne cycloaddition". Inorg. Chim. Acta, 2019, 498.
DOI: 10.1016/j.ica.2019.119136
146: [Invited Quitel 2018 Issue] Johanna Camacho, Alvaro Muñoz-Castro. "Shielding Cone Behavior in the Spherically Aromatic He@C60 6-. Original of the Record for the Most Shielded Encapsulated 3He Nucleus and Comparison to He@C70 6-". J. Mol. Mod., 2019, 25, 322. DOI: 10.1007/s00894-019-4211-4
145: Nickolas D. Charistos, Alvaro Muñoz-Castro. "Double aromaticity of B40 Fullerene: Induced magnetic field analysis of π and σ delocalization in boron cavernous structure".
Phys. Chem. Chem. Phys., 2019, 21, 20232-20238.
DOI: 10.1039/C9CP04223G
144: Franck Gam, Samia Kahlal, Ramiro Arratia-Perez, Jean-Yves Saillard, Alvaro Muñoz-Castro. "Stabilizing heteroatom- centered 16- vertex group 11 tetrahedral architectures: Bonding and structural considerations towards versatile endohedral species".
Int. J. Quantum Chem., 2019, 119, e26038.
DOI: 10.1002/qua.26038
143: Amr A Attia, Adrian Branzanic, Alvaro Muñoz-Castro, Alexandru Lupan, R. Bruce King. "Cationic Gold Clusters with Eight Valence electrons: Possible Spherical Aromatic Systems with Sigma Holes". Phys. Chem. Chem. Phys., 2019, 21, 17779-17785.
DOI: 10.1039/C9CP03440D
142: Daniela Fonseca, Sandra M. Leal-Pinto, Martha V. Roa-Cordero, José D. Vargas, rika M. Moreno-Moreno, Mario A. Macías, Leopoldo Suescun, Alvaro Muñoz-Castro, John J. Hurtado. "Inhibition of C. albicans Dimorphic Switch by Cobalt(II) Complexes with Ligans Derived from Pyrazoles and Dinitrobenzoate: Synthesis, Characterization and Biological Activity".
Int. J. Mol. Sci., 2019, 20, 3237.
DOI: 10.3390/ijms20133237
141: Shinji Toyota, Yuta Yamamoto, Kan Wakamatsu, Eiji Tsurumaki, Alvaro Muñoz-Castro. "Nano-Saturn with an Ellipsoidal Body: Anthracene Macrocyclic Ring-C70 Complex."
Bull. Chem. Soc. Jpn, 2019, 92, 1721-1728.
140: Alvaro Muñoz-Castro. "Potential of N-Heterocyclic Carbene Derivatives from Au13(dppe)5Cl2 Gold Superatoms. Evaluation of Electronic, Optical and Chiropatical Properties from Relativistic DFT." Inorg. Chem. Front, 2019, 6, 2349-2358.
DOI: 10.1039/C9QI00513G
139: [Backcover] Alvaro Muñoz-Castro. "Single, Double and Triple Intercluster Bonds. Analyses of M2Au36(SR)24 (M = Au, Pd, Pt) as 14-, 12- and 10-ve Superatomic Molecules."
Chem. Comm., 2019, 55, 7307-7310.
DOI: 10.1039/C9CC02970B
138: Alvaro Muñoz-Castro. "On the Ligand-Core Interaction in Ligand-Protected Gold Superatoms. Insights from Au(XR)18 ( = S, Se, Te) via Relativistic DFT Calculations."
Phys. Chem. Chem. Phys., 2019, 21, 13022-13029.
DOI: 10.1039/C9CP02077B
137: Mesias Orozco-Ic, Albeiro Restrepo, Alvaro Muñoz-Castro, Gabriel Merino. "Molecular Helmholtz coils". J. Chem. Phys., 2019. 151, 014102.
DOI: 10.1063/1.5094547
136: [Cover] Devaborniny Parasar, Naleen B. Jayaratna, Alvaro Muñoz-Castro, Allison E. Conway, Pavel K. Mykhailiuk, H. V. Rasika Dias. "Carbonyl complexes of copper (I) stabilized by bridging fluorinated pyrazolates and halide ions". Dalton Trans., 2019, 48, 6358-6371.
DOI: 10.1039/C9DT00486F
135: Nickolas D Charistos, Alvaro Muñoz-Castro, M.P. Sigalas. "The pseudo-π model of the Induced Magnetic Field: Fast and Accurate Visualization of Shielding and Deshielding Cones in Planar Conjugated Hydrocarbons and Spherical Fullerenes". Phys. Chem. Chem. Phys., 2019, 21, 6150-6159.
DOI: 10.1039/C9CP00836E
134: Ximena Zarate, Desmond MacLeod Carey, Alvaro Muñoz-Castro, Eduardo Schott. "Understanding the Aromaticity of C6X6 (X = H, F, Cl, Br, I). Insights from Different Theoretical Criteria". Chem. Phys. Lett., 2019, 720, 52-57.
DOI: 10.1016/j.cplett.2019.01.058
133: [Book Chapter] Alvaro Muñoz-Castro, Dayan Paez-Hernandez, Ramiro Arratia-Perez. "Rhenium hexanuclear clusters: Bonding, Spectroscopy and Applications of Molecular Chevrel Phases.". Struct. & Bond, 2019, 1-15.
DOI: 10.1007/430_2019_34
132: Franck Gam, Samia Kahlal, Ramiro Arratia-Perez, Jean- Yves Saillard, Alvaro Muñoz-Castro. "Potential to Stabilize 16-vertex Tetrahedral Coinage-Metal Cluster Architectures Related to that of Au20." Phys. Chem. Chem. Phys, 2019, 21, 8428-8433.
DOI: 10.1039/C9CP00639G
131: Nickolas D. Charistos, Peng Jin, Alvaro Muñoz-Castro. "Aromatic Character of Oh-C24-N24. A Cavernous Nitride Fullerene Bearing N4-Macrocycle Motifs."
J. Quant. Chem, 2019, 119, e25919.
DOI: 10.1002/qua.25919
130: Alan Miralrio, Alvaro Muñoz-Castro, R. Bruce King, Luis enrique Sansores. "M@C50 as Higher Intermediates towards Large Endohedral Metallofullerenes: Theoretical Characterization, Aromatic and Bonding Properties from Relativistic DFT Calculations."
J. Phys. Chem. C, 2019, 123, 2, 1429–1443.
129: Pengfei Ai, Matteo Mauro, Andreas A. Danopoulos, Alvaro Muñoz-Castro, Pierre Braunstein. "Dual Emission of a Cyclic Hexanuclear Gold(I) Complex. Interplay between Au3 and Au2 Ligand-Supported Luminophores." J. Phys. Chem C, 2019, 123, 915.
128: Franck Gam, Ramiro Arratia-Perez, Samia Kahlal, Jean-Yves Saillard, Alvaro Muñoz-Castro. "Symmetry lowering by cage doping in spherical superatoms: Evaluation of electronic and optical properties of 18-electron W@Au12Ptn (n= 0-4) superatomic clusters from relativistic DFT calculations." Int. J. Quant. Chem, 2019, e25827.
DOI: 10.1002/qua.25827
127: Alexander O. Ortolan, Nicholas D. Charistos, Raul Guajardo-Maturana, Carolina Olea Ulloa, Giovanni F. Caramori, Renato L. T. Parreira, Alvaro Muñoz-Castro. "On the cation-π capabilities of small all sp2- carbon host structure. Evaluation of [6.8]3cyclacene from relativistic DFT calculations." Int. J. Quant. Chem, 2019, 114, e25811.
DOI: 10.1002/qua.25811
126: [Review] Desmond Macleod-Carey, Giovanni F. Caramori, Raul Guajardo Maturana, Dayan Paez- Hernandez, Alvaro Muñoz-Castro and Ramiro arratia-Perez. "Advances in Bonding and Properties of Inorganic systems from Relativistic Calculations in Latin America"
Int. J. Quant. Chem, 2019, 119, e25777.
DOI: 10.1002/qua.25777
125: [Review] Alfredo Tlahuice-Flores, Alvaro Muñoz-Castro. "Bonding and Properties of Superatoms. Analogs to Atom and Moleculs and Related Concepts From Superatomic Clusters." Int. J. Quant. Chem, 2019, 119, e25756.
DOI: 10.1002/qua.25756
124: Gabriel Merino, Alvaro Muñoz-Castro, Marco Nascimiento, Alberto Vela. "Theoretical chemistry in Latin America" . Int. J. Quant. Chem, 2019, 119, e25756.
DOI: 10.1002/qua.25852
123: Alan Miralrio, Luis E. Sansores, R. Bruce King, Alvaro Muñoz-Castro. "C50Cl10, a planar aromatic fullerene. Computational study of 13C-NMR chemical shift anisotropy patterns and aromatic properties" Phys, chem. chem. Phys., 2018, 20, 26325.
DOI: 10.1039/C8CP04938F
122: Carolina Olea Ulloa, Miguel Ponce-Vargas, Alvaro Muñoz-Castro. "Formation of Coinage-Metal···Fullerene Adducts. Evaluation of the Interaction Nature between Triangular Coinage Metal Complexes (M3 = Cu, Ag and Au) and C60 through Relativistic Density Functional Theory Calculations." J. Phys. Chem. C, 2018, 122, 25110.
121: Carolina Olea Ulloa, Miguel Ponce-Vargas, Alvaro Muñoz-Castro. "Nature of cucurbituril-halogen encapsulation. Structural and interaction energy consideration in the X2@CB[n] (X= Cl, Br, I, n = 6, 7, 8) from relativistic DFT calculations", Phys. Chem, Chem. Phys., 2018, 20, 29325.
DOI: 10.1039/C8CP04936J
120: Ricardo A. Murcia, Sandra M. Leal, Martha V. Roa, Edgar Nagles, Alvaro Muñoz-Castro, John J. Hurtado. "Development of Antibacterial and Antifungal Triazole Chromium (III) and Cobalt (II) Complexes: Synthesis and Biological Activity Evaluations", Molecules, 2018, 23, 2013.
DOI: 10.3390/molecules23082013
119: Sukanta Mondal, Pallavi Sarkar, Alvaro Muñoz-Castro. "Planar ten-membered 10-π-electron aromatic (CH)5(XH)5 {X=Ge, Sn} systems". J. Mol Model, 2018, 24, 264.
DOI: 10.1007/s00894-018-3797-2
118. Juan F. Torres, Nestor J. Bello-Vieda, Mario A. Macías, Alvaro Muñoz-Castro, Carlos Rojas-Dotti, José Martínez-Lillo, John Hurtado. Water Dissociation of a Dinuclear bis(3,5‐dimethylpyrazolyl)methane Copper(II) Complex. X‐ray Structure, Magnetic Properties and Characteristic Absorption of the (CuN2Cl2)2 Core. Eur. J. Inorg. Chem. 2018, 37, 3644-361.
117: Alexandre O. Ortolan, Giovanni F. Caramori, Renato L. T. Parreira, Alvaro Muñoz-Castro. "Helicenes as Molecular Tweezers in the Formation of Cation-p Complexes. Bonding and Circular Dichroism Properties from Relativistic DFT Calculations".
Chem Phys Chem., 2018, 19, 2321 –2330.
116: [Review] Alfredo Tlahuice-Flores, Alvaro Muñoz-Castro. "Bonding and Properties of Superatoms. Analogs to Atoms and Molecules and Related Concepts From Superatomic Clusters". Int. J. Quantum Chem., 2018, 118, e25508.
DOI: 10.1002/qua.25508
115: Miguel Ponce-Vargas and Alvaro Muñoz-Castro. "THE STABILIZING ROLE OF HALIDE IONS IN ENDOHEDRAL [20]SILAFULLERANES: INSIGHTS FROM DFT CALCULATIONS TOWARDS SILICON NANOCAGES". J. Phys. Chem. C, 2018, 122, 23, 12551–12558.
114. Alvaro Muñoz-Castro and Jean-Yves Saillard. [Au12(SR)6]2‐, As Smaller 8‐electron Gold Nanocluster Retaining an SP3‐core. Evaluation of Bonding and Optical Properties from Relativistic DFT Calculations. Chem Phys Chem., 2018, 19, 1846-1851.
113. Desmond MacLeod Carey and Alvaro Muñoz-Castro, Au11Re: A Hollow or Endohedral Binary Cluster?. Chem.. Phys. Lett. 2018, 701, 30-33.
DOI: 10.1016/j.cplett.2018.04.038
112. Nickolas D. Charistos, Alvaro Muñoz-Castro. Induced Magnetic Field of Fullerenes. Role of - and - contributions to Spherical Aromatic, Non-Aromatic and Antiaromatic Character in C60q (q = +10, 0, -6, -12), and Related Alkali-Metal Decorated Building Blocks, Li12C60 and Na6C60. J. Phys. Chem. C, 2018. 122, 9688-9698.
111: Francisca Claveria-Cadiz, Ramiro Arratia-Perez, Raúl Guajardo-Maturana, Alvaro Muñoz-Castro. "Survey of Short and Long Cuprophilic d10-d10 Contacts for Tetranuclear Copper Clusters. Understanding of Bonding and Ligand Role from Planar Superatom Perspective".
New J. Chem., 2018, 42, 8874-8881.
DOI: 10.1039/c8nj00698a
110. Franck Gam, Ramiro Arratia-Pérez, Samia Kahlal, Jean-Yves Saillard and Alvaro Muñoz-Castro. [M16Ni24(CO)40]4-: "Coinage Metal Tetrahedral Superatoms as Useful Building Blocks Related to Pyramidal Au20 cluster (M = Cu, Ag, Au). Electronic and Bonding Properties from Relativistic DFT Calculations". J. Phys. Chem. C, 2018, 122,4723-4730.
109. Nestor J. Bello-Vieda, Homero F. Pastrana, Manuel F. Garavito, Alba G. Ávila, Adriana M. Celis, Alvaro Muñoz-Castro, Silvia Restrepo and John J. Hurtado. "Antibacterial Activities of Azole Complexes Combined with Silver Nanoparticles." Molecules 2018, 23, 361.
DOI: 10.3390/molecules23020361
108. Alvaro Muñoz-Castro. "Local and Global Aromaticity in a Molecular Carbon Nanobelt. Insights from Magnetic Response Properties in Neutral and Charged Species"
Phys. Chem. Chem. Phys. 2018,20, 3433-3437.
DOI: 10.1039/C7CP08323H
107. Glaucio R. Nagurniak, Giovanni F. Caramori, Alvaro Muñoz-Castro, Renato L. Perreira, Éder H. da Silva, "The Ability of Ex2Box4+ to Interact with Guests Containing π-Electron-Rich and π-Electron-Poor Moieties". Int J Quantum Chem., 2018, 118, e25607.
DOI: 10.1002/qua.25607
106. Alvaro Muñoz-Castro, "Fulfilling the 2(N+1)2 Hirsch rule in Smaller Hollow Fullerenes. Evaluation of Long-Range Magnetic Behavior and NMR Patterns of C28, C284-, C24N4 and C28H4". Int. J. Quant. Chem., 2018, 113, e25645.
DOI: 10.1002/qua.25645
105. Alan Miralrio, Alvaro Muñoz-Castro, R. Bruce King, Luis Enrique, "Sansores. On the Intermediates for Larger Endohedral Metallofullerenes: Theoretical Characterization of M@C44 Species". J. Phys. Chem. C.,2018, 122, 798–807.
104. Nestor J. Bello-Vieda, Ricardo A. Murcia, Alvaro Muñoz-Castro, Mario A. Macías, John J. Hurtado. "Metals complexes containing 1,3-phenylenebis((1H-1,2,4-triazol-1-yl)methanone) ligands: Synthesis and є-caprolactone polymerization behavior". Molecules 2017, 22, 1860.
DOI: 10.3390/molecules22111860
103. Nuñez-Dallos, Natalia Lopez-Barbosa, Alvaro Muñoz-Castro, Desmond Mac-Leod Carey, Assunta De Nisi, Magda Monari, Johann F. Osma and John Hurtado. "A new Copper(I) coordination polymer from 2,6-bis(1H-benzotriazol-1-ylmethyl)pyridine: synthesis, characterization, and use as additive in transparent submicron UV filters".
J. Coord. Chem. 2017, 70, 19, 3363–3378.
DOI: 10.1080/00958972.2017.1393072
102. Alvaro Muñoz-Castro, R. Bruce King, Formation of Spherical Aromatic Endohedral Metallic Fullerenes. Evaluation of Magnetic Properties of M@C28 (M = Ti, Zr and Hf) from DFT calculations. Inorg. Chem., 2017, 56, 15251-15258.
DOI: 10.1021/acs.inorgchem.7b02611
101. Fahri Alkan, Alvaro Muñoz-Castro, "Christine Aikens, Relativistic DFT Investigation of Electronic Structure Effects Arising from Doping the Au25 Nanocluster with Transition Metals".
Nanoscale, 2017, 9, 15825-15834.
DOI: 10.1039/C7NR05214F
100. Luis G. Perla, Alvaro Muñoz-Castro, and Slavi C. Sevov. "Eclipsed- and Staggered-[Ge18Pd3{EiPr3}6]2– (E = Si, Sn): Positional Isomerism in Deltahedral Zintl Clusters",
J. Am. Chem. Soc. 2017, 139, 15176-15181.
DOI: 10.1021/jacs.7b08562
99. Alvaro Muñoz-Castro. "A Superatomic Molecule under the Spin-Orbit Coupling. Insights from the Electronic Properties in the Thiolate-Protected Au38(SR)24 Cluster". Int J Quantum Chem. 2018, 118, e25508.
DOI: 10.1002/qua.25508
98. Alvaro Muñoz-Castro, Ivan A. Popov and Alexander Boldyrev. "Long-Range Magnetic Response of Toroidal Boron Structures: B16 and [Co@B16]−/3− Species". Phys. Chem. Chem. Phys., 2017,19, 26145-26150.
DOI: 10.1039/C7CP04158F
97. Alexandre O. Ortolan, Giovanni F. Caramori, F. Matthias Bickelhaupt, Renato L. T. Parreira, Alvaro Muñoz-Castro and Tapas Kar. " How the Electron-Deficient Cavity of Heterocalixarenes Recognizes Anions. Insights from Computation". Phys. Chem. Chem. Phys., 2017,19, 24696-24705.
DOI: 10.1039/C7CP03925E
96. Nickolas D. Charistos, Anastasios G. Papadopoulos, Thomas A. Nikopoulos, Alvaro Muñoz-Castro, Michael P. Sigalas. "Canonical Orbital Contributions to the Magnetic Fields Induced by Global and Local Diatropic and Paratropic Ring Currents". Journal of Computational Chemistry 2017, 38, 2594–2604.
DOI: 10.1002/jcc.24917
95. Franck Gam, Dayan Paez-Hernandez, Ramiro Arratia-Perez, C. W. Liu, Samia Kahlal, Jean-Yves Saillard, A. Muñoz-Castro. "Coinage Metal Superatomic Cores. Insights into their Intrinsic Stability and Optical Properties from Relativistic DFT Calculations".
Chem. Eur. J. 2017, 23, 11330-11337.
94. F. Murillo, A. Vargas-Caamal, S. Pan, J. L. Cabellos, M. Mora-Fonz, A. Muñoz-Castro, A. Restrepo, G. Merino. Does H4SO5 exist?. Phys. Chem. Chem. Phys., 2017,19, 17088-17093.
DOI: 10.1039/C7CP01328K
93. J. Hurtado, L. Ibarra, D. Yepes, P. García-Huertas, M. A. Macías, O. Triana-Chavez, E. Nagles, L. Suescun, A. Muñoz-Castro. Synthesis, Crystal Structure, Catalytic and Anti-Trypanosoma cruzi Activity of a New Chromium(III) Complex Containing Bis(3,5-dimethylpyrazol-1-yl)methane.
J. Mol. Str. 1146, 2017, 365e372.
DOI: 10.1016/j.molstruc.2017.06.014
92. Alvaro Muñoz-Castro and R. Bruce King. Aromatic and Antiaromatic Spherical Structures: Use of Long-Range Magnetic Behavior as an Aromatic Indicator for Bare Icosahedral [Al@Al12]– and [Si12]2– Phys. Chem. Chem. Phys., 2017,19, 15667-15670.
DOI: 10.1039/C7CP02607B
91. Alvaro Muñoz-Castro. Shielding Cone in Spherical Aromatic Structures. Insights from Models for Spherical 2(N+1)2 Aromatic Fullerenes. Phys. Chem. Chem. Phys., 2017,19, 12633-12636.
DOI: 10.1039/C7CP01870C
90. Vanessa Molina, Markus Rauhalahti, John Hurtado, Heike Fliegl, Dage Sundholm and Alvaro Muñoz-Castro. "Aromaticity Introduced by Antiferromagnetic Ligand Mediated Metal-Metal Interactions. Insights from the Induced Magnetic Response in [Cu6(dmPz)6(OH)6]".
Inorg. Chem. Front., 2017,4, 986-993.
DOI: 10.1039/C7QI00023E
89. A. G. Papadopoulos, N. D. Charistos and A. Muñoz-Castro. "Magnetic Response of Aromatic Rings Under Rotation. Aromatic Shielding Cone of Benzene Upon Different Orientations of the Magnetic Field". ChemPhysChem 2017, 18,1499 –1502.
88. Alvaro Muñoz-Castro, Wilson Caimanque-Aguilar, and Cesar Morales-Verdejo, "Computational Study of 13C NMR Chemical Shift Anisotropy Patterns in C20H10 and [C20H10]4–. Insights into Their Variation upon Planarization and Formation of Concentric Aromatic Species in the Smaller Isolated-Pentagon Structural Motif". J. Phys. Chem. A 2017, 121, 13, 2698–2703.
87. Alvaro Muñoz-Castro and R. Bruce King. "On the Formation of Smaller p-Block Endohedral Fullerenes: Bonding Analysis in the E@C20 (E = Si, Ge, Sn, Pb) Series from Relativistic DFT Calculations". Journal of Computational Chemistry 2017, 38, 1661–1667.
DOI: 10.1002/jcc.24809
86. Alvaro Muñoz-Castro and R. Bruce King. "Au20. Effect of a Strong Tetrahedral Field in a Spherical Concentric Bonding Shell Model". J. Phys. Chem. C. 2017, 121, 10, 5848–5853.
85. [Invited Ahmed H. Zewail Special Issue] Dayan Paez-Hernandez, Alvaro Muñoz-Castro, Ramiro Arratia-Perez. "Bonding in Gold-Rare Earth [Au2M] (M = Eu, Yb, Lu) ions. A Strong Covalent Gold-Lanthanide Bond" Chem. Phys. Lett. 2017, 684, 421-424.
DOI: 10.1016/j.cplett.2017.03.074
84. [Invited Ahmed H. Zewail Special Issue] Alvaro Muñoz-Castro, Dayan Paez-Hernandez, Ramiro Arratia-Perez. Spin-Orbit Effect into Isomerization Barrier of Small Gold Clusters. Oh↔D2h Fluxionality of the Au62+ Cluster Investigated by Relativistic Methods. Chem. Phys. Lett.,2017, 683, 404-407.
DOI: 10.1016/j.cplett.2017.02.054
83. Alvaro Muñoz-Castro and Keisuke Takahashi. "Towards Two-Dimensional Superatomic Honeycomb Structure. Evaluation of [Ge9(Si(SiMe3))3]- as Source of Ge9-Cluster Building Blocks for Extended Materials".J. Phys. Chem. C, 2017, 121, 1934–1940.
82. Alvaro Muñoz-Castro. "Doping the Cage. Re@Au11Pt and Ta@Au11Hg, As Novel 18-ve Trimetallic Superatoms Displaying a Doped Icosahedral Golden Cage".
Phys. Chem. Chem. Phys., 2017,19, 2459-2465.
DOI: 10.1039/C6CP07519C
81. Alvaro Muñoz-Castro and R. Bruce King . "Au102+ and Au6X42+ clusters: Superatomic molecules bearing an sp3-hybrid Au6 core". Int. J. Quant. Chem. 2017, 117, e25331.
DOI: 10.1002/qua.25331
80. Alvaro Muñoz-Castro. "Evaluation of Hollow Golden Icosahedrons. Bonding and Spherical Aromatic Properties of [Au11E]3- Superatoms (E= Se and Te) from Relativistic DFT calculations, Persistent Structures?". Chem. Phys. Chem., 2017, 18, 87–92.
79. Alvaro Muñoz-Castro and R. Bruce King. "Evaluation of Bonding, Electron Affinity, and Optical Properties of M@C28 (M= Zr, Hf, Th, and U): Role of d- and f-Orbitals in Endohedral Fullerenes from Relativistic DFT Calculations".
J. Compt. Chem. 2017, 38, 44–50.
DOI: 10.1002/jcc.24518
78. Fernando Mendizabal, Sebastian Esteban Miranda-Rojas, Alvaro Muñoz-Castro, Ramiro Arratia, Jose Zagal and Paula Sierra-Rosales. "Catalytic Aspects of Metallophtalocyanines Adsorbed on Gold‑Electrode. Theoretical Exploration of the Binding Nature Role".
Phys. Chem. Chem. Phys., 2016, 18, 29516-29525.
DOI: 10.1039/C6CP06156G
77. Alvaro Muñoz-Castro. "D6h-Au42 Isomer: A Golden Aromatic Toroid Involving Superatomic π-Orbitals that Follow the Hückel (4n+2)π rule". ChemPhysChem, 2016, 17, 3204–3208.
76. Alvaro Muñoz-Castro. "U@C36. Is there enough room for a second uranium?". RSC Adv., 2016, 6, 78176-78180.
DOI: 10.1039/C6RA15471A
75. Miguel Ponce-Vargas and Alvaro Muñoz-Castro. "Tiara-like Complexes acting as Iodine Encapsulating Agents: The Role of M···I Interactions in [M(μ-SCH2CO2Me)2]8⊂I2 (M = Ni, Pd, Pt) Inclusion Compounds". J. Phys. Chem. C, 2016, 120, 23441–23448.
74. Johanna Camacho Gonzalez and Alvaro Muñoz-Castro, "Doping the superatom with p-elements. Role of p-block Endohedral atoms in Bonding and Optical Properties of E@Au24(SR)18 (E= Si, Ge, Sn and Pb) from Relativistic DFT calculations". J. Phys. Chem. C, 2016, 120, 27019-27026.
73. Glaucio R. Nagurniak, Giovanni F. Caramori, Renato L. T. Parreira, Pedro A. S. Bergamo, Gernot Frenking, and Alvaro Muñoz-Castro. "Shedding Light on the Nature of Host–Guest Interactions in PAHs-ExBox4+ Complexes". J. Phys. Chem. C, 2016, 120, 15480–15487.
72. Alvaro Muñoz-Castro, Markus Rauhalahti and Dage Sundholm, "Magnetic Response Properties of Gaudiene - A Cavernous and Aromatic Carbocage", Phys. Chem. Chem. Phys., 2016, 18, 18880-18886.
DOI: 10.1039/C6CP03808E
71. Feng Li, Alvaro Muñoz-Castro, Slavi C. Sevov. "[(Me3Si)Si]3EtGe9Pd(PPh3), a Pentafunctionalized Deltahedral Zintl Cluster: Synthesis, Structure, and Solution Dynamics." Angew. Chem. Int. Ed. 2016, 55, 8630–8633.
70. Alvaro Muñoz-Castro. "The impact of endohedral atoms on the electronic and optical properties of Au25(SR)18 and Au38(SR)24". Phys. Chem. Chem. Phys., 2016,18, 31419-31423. DOI: 10.1039/C6CP02691E
69. Sebastián Miranda-Rojas, Alvaro Muñoz-Castro, Ramiro Arratia-Pérez ,Fernando Mendizábal
"Theoretical Aspects of the Reactivity of MN4 Macrocyclics in Electrochemical Reactions", Springer International Publishing 2016, 1, 143-170.
DOI: 10.1007/978-3-319-31172-2_5
68. Anastasios G. Papadopoulos, Nickolas D. Charistos ,Alvaro Muñoz-Castro
"On the role of heteroatoms in aromatic rings. Insights from 10p main group elements heterorings [(EH)2S2N4] q (E = C, P, B, Si, Al and q = 0, 2)". New J. Chem., 2016, 40, 5090-5098.
DOI: 10.1039/C5NJ03573B
67. Alvaro Muñoz-Castro, Desmond MacLeod-Carey, Sebastian Miranda-Rojas, Fernando Mendizabal, Jose H. Zagal and Ramiro Arratia-PÉrez "Surface on Surface. Survey of the Monolayer Gold–Graphene Interaction from Au12 and PAH via Relativistic DFT Calculations"
J. Phys. Chem. C, 2016, 120 (13), 7358–7364.
66. R. Guajardo Maturana, A. Muñoz-Castro. "Insights into metal–ligand and metal–metal interaction in coinage metal triangles. Insights of d10-d10, d10-d8 and d8-d8 contacts from [Au3In(CH3N COCH3)3] (n = 2, 4, 6) via relativistic DFT calculations" Chem. Phys.Lett., 2016, 651,34–38.
DOI:10.1016/j.cplett.2016.03.013
65. Alvaro Muñoz-Castro "On the fused-to-single ring transition in 10π structures. Insights from naphthalene to [10]annulene series". Chem. Phys. Lett., 2016, 650, 16 , 60–63.
DOI: 10.1016/j.cplett.2016.02.064
64. M. Rauhalahti, A. Muñoz-Castro, D. Sundholm. "Thiolate-protected golden fullerenes. A 32-ve core involving a hollow Au32 cage". RSC Adv., 2016,6, 21332-21336.
DOI: 10.1039/C5RA27683G
63. Alexandre O. Ortolan, Giovanni F. Caramori, Gernot Frenking , Alvaro Muñoz-Castro
"Role of the cation formal charge in cation–p interaction. A survey involving the [2.2.2]paracyclophane host from relativistic DFT calculations"
New J. Chem., 2015,39, 9963-9968.
DOI: 10.1039/C5NJ02384J
62. K. F. Andriani,G. F. Caramori, A. Muñoz-Castro, F. G. Doro, "The Influence of L Ligands on the {RuNO}6/7 Bonding Situation in cis-[Ru(NO)(NO2)L1-4]q Complexes: A Theoretical Insight".
RSC Adv., 2015, 5, 69057-69066.
DOI: 10.1039/C5RA10888H
61. M. Ponce Vargas A. Muñoz-Castro, "Metal Containing Cryptands as Hosts for Anions. Evaluation of Cu(I)•••X and p•••X interactions in Halide-Tricopper(I) Complexes through Relativistic DFT Calculations.". Phys. Chem. Chem. Phys., 2015, 2015,17, 18677-18683.
DOI: 10.1039/C5CP02737C
60. J. Camacho-Gonzalez, A. Muñoz-Castro, "Alternation of Aromatic-Nonaromatic Rings in Belt-like Structures. Behavior of [6.8]3cyclacene in Magnetic Fields.".
Phys. Chem. Chem. Phys., 2015, 17, 17023-17026.
DOI: 10.1039/C5CP02195B
59. D. MacLeod Carey,T. Gomez, C. Morales-Verdejo, A. Muñoz-Castro, "Influence of Ag+ on the Magnetic Response of [2.2.2]Paracyclophane: NMR properties of a Prototypical Organic Host for Cation Binding Based on DFT calculations". Chem.Open 2015, 4, 651-655.
58. A. Muñoz-Castro, "Axis-dependent Magnetic Behavior of C60 and C6010+. Consequences of Spherical Aromatic Character.". Chem. Comm., 2015, 51, 10287-10290.
DOI: 10.1039/C5CC03352G
57. J. Camacho Gonzalez, C. Morales Verdejo, A. Muñoz-Castro, “Variation of trough-space magnetic response properties upon formation of Cation-π interactions. A survey of [Ag(η-CH2CH2)3]+ via DFT calculations”. New J. Chem. 2015, 39, 4244-4248.
DOI: 10.1039/C5NJ00475F
56. D. MacLeod Carey, C. Morales-Verdejo, A. Muñoz-Castro, “[As@ Ni 12@ As 20] 3− and [Sn@ Cu 12@ Sn 20] 12− clusters. Related structures with different construction philosophy”, Chem. Phys. Lett. 2015, 638, 99-102.
DOI: 10.1016/j.cplett.2015.08.039
55. J. Camacho Gonzalez, A. Muñoz-Castro, “WOMAN IN CHEMISTRY. JANE MARCET, A RELEVANT FIGURE IN CHEMISTRY EDUCATION” , Química Nova,38, 1374-1378 2015.
DOI: 10.5935/0100-4042.20150143
54. M. Rauhalahti, A. Muñoz-Castro, "Interaction in Multilayer Clusters: A Theoretical Survey of [Sn@Cu12@Sn20]12-, a Three-layer Matryoshka-like Intermetalloid"., RSC Advances, 2015, 5, 18782-18787.
DOI: 10.1039/C4RA16660D
53. C. Olea Ulloa, M. Ponce-Vargas, R. M. Piccoli, G. F. Caramori, G. Frenking, A. Muñoz-Castro, "[2.2.2]Paracyclophane, Preference for η6 or η18 Coordination Mode Including Ag(I) and Sn(II): A Survey into the Cation-π Interaction Nature Through Relativistic DFT Calculations."., RSC Advances, 2015,5, 7803-7811.
DOI: 10.1039/C4RA12859A
52. M. Ponce-Vargas, A. Muñoz-Castro, "Heavy Elements Metallacycles: Insights Into the Nature of Host-Guest Interactions Involving Di-Halide Mercuramacrocycle Complexes"., J. Phys. Chem. C, 2014, 118, 28244–28251.
DOI: 10.1021/jp5092625
51. G. F. Caramori, R. M. Piccoli, M. Segala, A. Muñoz-Castro, R. Guajardo-Maturana, D. M. Andrada, G. Frenking "Cyclic Trinuclear Copper(I), Silver(I), and Gold(I) Complexes: A Theoretical Insight.", Dalton Trans. 2014, 44, 377-385.
DOI: 10.1039/C4DT02514H
50. A. Muñoz-Castro, "sp3-hybridization in superatomic clusters. Analogues to simple molecules involving the Au6 core." Chem. Sci. 2014, 5, 4749-4754.
DOI: 10.1039/C4SC01719F
49. R. Guajardo Maturana, A. Muñoz-Castro, "Understanding Planar Ligand-Supported MAu5 and MAu6 cores. Theoretical Survey of [MAu5(Mes)5] and [MAu6(Mes)6] (M=Cu, Ag, Au; Mes=2,4,6-Me3C6H2) Under The Planar Superatom Model." J. Phys. Chem. C 2014, 118, 21185–21191.
DOI: 10.1021/jp5057557
48. M. Ponce-Vargas, A. Muñoz-Castro, "On the versatility of metallacycles in Host-Guest Chemistry. Interactions in Halide-centered hexanuclear copper(II) pyrazolate complexes", Phys. Chem. Chem. Phys. 2014, 16, 13103-13111.
DOI: 10.1039/C4CP01238K
47. A. Muñoz-Castro, "Application of planar superatom model on [Hg5(C(CF3)2)]. Bonding and magnetic response considerations into a five-fold d10-d10 metal cycle", Phys. Chem. Chem. Phys. 2014,16, 7578-7583.
DOI: 10.1039/C3CP55482A
46. C.Morales-Verdejo, I. Martínez-Díaz, C. Adams, J. F. Araneda, L. Oehninger, D. Mac-Leod Carey, A. Muñoz-Castro, R. Arratia-Pérez, I. Chávez, J. M. Manríquez, "New mono and bimetallic iron complexes derived from partially methylated s-indacene. Evidence of a trinuclear iron s-indacene complex". Polyhedron, 2014, 69, 15-24.
DOI: 10.1016/j.poly.2013.11.023
45. A. Muñoz-Castro, "Golden Endohedral Main-Group Clusters, [E@Au12]q-: Theoretical Insights Into the 20-e Principle", J. Phys. Chem. Lett, 2013, 4, 3363-3366.
DOI: 10.1021/jz401622m
44. S. Miranda-Rojas, A. Muñoz-Castro, R. Arratia-Pérez, F. Mendizábal "Insights into the Adsorption of Neutral, Radical and Anionic Thiophenols on Cluster of Gold (111)", Phys. Chem. Chem. Phys., 2013, 15, 20363-20370.
DOI: 10.1039/C3CP53591F
43. L. Oehninger, N. Küster, C. Schmidt, A. Muñoz-Castro, A. Prokop, I. Ott, "A Chemical-Biological Evaluation of Rhodium(I) N-Heterocyclic Carbene Complexes as Prospective Anticancer Drug", Chem. Eur. J. 2013, 19, 17871-17880.
42. F. Ferraro, D. Páez Hernández, J. Murillo-Lopez, A. Muñoz-Castro, R. Arratia-Perez, "Antenna Effect by Organometallic Chromophores in Bimetallic d-f Complexes", J. Phys. Chem. A, 2013, 117, 7847-7854.
DOI: 10.1021/jp406208e
41. C. Olea Ulloa, M. Ponce Vargas, R. Guajardo Maturana, A. Muñoz-Castro, "Theoretical Study of the binding strength and magnetical response properties involved in the formation of the p-donor/p-acceptor [TTF-CBPQT]4+ host-guest system", Polyhedron, 2013, 54, 119-122.
DOI: 10.1016/j.poly.2013.02.022
40. F. MIRANDA-BARRIENTOS, A. MUNOZ-CASTRO, R. ARRATIA-PEREZ, D. MACLEOD CAREY, "THEORETICAL CALCULATIONS OF AN OSMIUM MOLECULAR SWITCH". J. Chil. Chem. Soc. 2013, 58, 2110-2113.
DOI: 10.4067/S0717-97072013000400046
39. A. Muñoz-Castro, S. C. Sevov "Tri-metallic deltahedral Zintl ions [Sn9-m-nGemBin](4-n)- for n = 1 – 4 and m = 0 – (9-n): A theoretical survey with prediction and rationalization of the possible structures", Phys. Chem. Chem. Phys., 2013,15, 986-991.
DOI: 10.1039/C2CP43196C
38. L. Oehninger, M. Stefanopoulou, H. Alborzinia, J. Schur, S. Ludewig, K. Namikawa, A. Muñoz-Castro, R. W. Köster, K. Baumann , S. Wölfl , W. S. Sheldrick, I. Ott, "Evaluation of Arene Ruthenium(II) N-heterocyclic Carbene Complexes as Organometallics Interacting with Thiol and Selenol Containing Biomolecules.", Dalton Trans., 2013,42, 1657-1666.
DOI: 10.1039/C2DT32319B
37. L. Oehninger, J. F. Araneda, C. Morales-Verdejo, A. Muñoz-Castro, D. Mac-Leod Carey, C. Adams, R. Arratia-Pérez, R. Rojas, J. M. Manríquez, I. Chávez, "Novel Titanocene derived from a partially alkylated s-indacene: Synthesis, characterization and comparative study with its zirconium analog.", Inorg. Chim. Acta., 2013, 396, 35-39.
DOI: 10.1016/j.ica.2012.09.032
36. A. Muñoz-Castro, "On the Magnetic Behavior of Spherical Aromatic compounds. Insights from the closo-[B12H12]2- cluster through chemical shift tensor maps.", Chem. Phys. Lett. 2013, 3, 282-285.
DOI: 10.1016/j.cplett.2012.10.083
35. F. Li, A. Muñoz-Castro, S. C. Sevov, "[Ge9{Si(SiMe3)3}3{SnPh3}]: A Neutral Tetra-Substituted Deltahedral Nine-Atom Cluster.", Angew. Chem. Int. Ed. 2012, 51, 8581-8584.
34. R. Guajardo Maturana, M. Ponce Vargas, A. Muñoz-Castro, "Survey of long d10-d10 metallophilic contacts in four membered rings of Ag(I) and Au(I) supported by carbene-pyrazole mixed ligands." J. Phys. Chem. A, 2012, 116, 8737-8743.
DOI: 10.1021/jp304928k
33. J. Camacho Gonzalez, A. Muñoz-Castro, "Survey of the Magnetic Behavior of [TTF] and [TTF2]2+ from Shielding Tensor Maps.", Chem. Phys. Lett. 2012, 543, 184-187.
DOI: 10.1016/j.cplett.2012.06.028
32. A. Muñoz-Castro, "Magnetic response properties of coinage metal macrocyles. Analysis of [Cu5(Mes)5], [Ag4(Mes)4] and [Au5(Mes)5] (Mes= 2,4,6-Me3C6H2)", J. Phys. Chem. C, 2012, 116, 17197-17203.
DOI: 10.1021/jp3012443
31. I. Ponce, J. F. Silva, R. Oñate, J. Pavez, J. H. Zagal, F. Mendizabal, S. Miranda-Rojas, A. Muñoz-Castro, R. Arratia-Pérez. "Enhancement of the Catalytic activity of Fe phthalocyanine for the reduction of O2 anchored to Au(111) via conjugated self-assembled monolayers of aromatic thiols compared to Cu phthalocyanine" , J. Phys. Chem. C 2012, 116, 15329-15341.
DOI: 10.1021/jp301093q
30. D. Mac-Leod Carey, C. Adams, n, C. Morales-Verdejo, J. F. Araneda, I. Chavez, J. M. Manríquez, A. Castel, P. Rivière, M. Rivière-Baudet, D. Matioszek, R. Septelean, I. Martinez, R. Arratia-Pérez. "A new method to radical anions derived from s-Indacene organobimetallic complexes, their ESR characterization",Inorg. Chim. Acta. 2012, 392, 154-159.
DOI: 10.1016/j.ica.2012.05.033
29. A. Muñoz-Castro, D. Mac-Leod Carey, R. Arratia-Perez, G. L. Malli, "Relativistic effects in bonding and isomerization energy of the superheavy roentgenium (111Rg) cyanide", Polyhedron, 2012, 39, 113-117.
DOI: 10.1016/j.poly.2012.03.032
28. C. Linares-Flores, D. Mac-Leod Carey, A. Muñoz-Castro, J. H. Zagal, J. Pavez, D. Pino-Riffo, and R. Arratia-Pérez. "Reinterpreting the Role of the Catalyst Formal Potential. The case of Thiocyanate Electrooxidation Catalyzed by CoN4-Macrocyclic Complexes.", J. Phys. Chem. C, 2012, 116, 7091-7098. DOI: 10.1021/jp300764n
27. Rohit Singh Chauhan, G. Kedarnath, Amey Wadawale, Arnold L. Rheingold, Alvaro Muñoz-Castro, Ramiro Arratia-Perez, and Vimal K. Jain, "Reactivity of Dipyridyl Ditellurides with (Diphosphine)Pt0 and 2-Pyridyltellurolates with (Diphosphine)PtCl2 and Isolation of Different Structural Motifs of Platinum(II) Complexes", Organometallics, 2012, 31, 1743-1750.
DOI: 10.1021/om2010589
26. M. M. Gillett-Kunnath, A. Muñoz-Castro, S. C. Sevov, “Tri-metallic deltahedral Zintl ions: Experimental and theoretical studies of the novel dimer [(Sn6Ge2Bi)2]4-“, Chem. Comm. 2012, 48, 3524-3526.
DOI: 10.1039/C2CC30459G
25. C. Echeverria, A. Becerra, F. Nuñez-Villena, A. Muñoz-Castro, J. Stehberg, Z. Zheng, R. Arratia-Perez, F. Simon, R. Ramirez-Tagle, “The paramagnetic and luminescent [Re6Se8I6]3- cluster. Its potential use as an antitumoral and biomarker agent.“
New J. Chem. 2012, 36, 927-932.
DOI: 10.1039/C2NJ21016A
24. A. Muñoz-Castro, R. Arratia-Pérez, “Spin-Orbit Effects on a Gold-Based Superatom: A Relativistic Jellium Model“, Phys. Chem. Chem. Phys., 2012, 14, 1408-1411
DOI: 10.1039/C1CP22420D
23. A. Muñoz-Castro, “On the Nature of the Excited State of Triangulo Silver N-Heterocyclic Carbenes. Insights from Relativistic DFT Calculations“, J. Phys. Chem. A. 2012, 116, 520-525.
DOI: 10.1021/jp2071277.
22. I. Ponce, J. Silva, R. Onate, S. Miranda-Rojas, A. Muñoz-Castro, R. Arratia-Perez, F. Mendizabal, J. Zagal, “Theoretical and Experimental Study of Bonding and Optical Properties of Self-Assembly Metallophthalocyanines Complexes on Gold Surface. A Survey of the Substrate-Surface Interaction“, J. Phys. Chem. C. 2011, 115, 23512-23518.
21. A. Muñoz-Castro, “Behavior of [2.2]Paracyclophane in Magnetic Fields. A Survey of the Magnetic Response Properties from Chemical Shift Tensor Maps“, Chem. Phys. Lett. 2011, 517, 113-115
DOI:10.1016/j.cplett.2011.10.028.
20. J. Hurtado, J, Ugarte, R. Rojas, M. Valderrama, D. Mac-Leod Carey, A. Muñoz-Castro, R. Arratia-Pérez, R. Fröhlich, “New bis(azolylcarbonyl)pyridine chromium(III) complexes as initiators for ethylene polymerization“, Inorg. Chim. Acta, 2011, 378, 218-223
DOI:10.1016/j.ica.2011.08.065.
19. A. Muñoz-Castro, “Ni2S2, a robust building block. Insights from relativistic DFT calculations in several toroid structures“, J. Phys. Chem. A, 2011, 115, 10789–10794.
DOI:10.1021/jp2028438.
18. L.-C.Pop, D. Mac-Leod Carey, A. Muñoz-Castro, L. Silaghi-Dumitrescu, A. Castel, R. Arratia-Pérez “Relativistic calculations of aminotroponiminate complexes containing group 15 (P, As, Sb, Bi) elements“, Polyhedron 2011, 30, 841-845
DOI:10.1016/j.poly.2010.12.023.
17. X. Tu, E. Alster, A. Muñoz-Castro, R. Arratia-Pérez, G. S. Nichol, Z. Zheng, “Geometrically Specific Imino Complexes of the [Re6(µ3-Se)8]2+ Core-containing Clusters“,Chem. Eur. J., 2011, 17, 580-587.
DOI: 10.1002/chem.201001975.
16. N.-D. Van, I. Tiritiris, R. F. Winter, B. Sarkar, P. Singh, C. Duboc, A. Muñoz-Castro, R. Arratia-Pérez, W. Kaim, T. Schleid “Hydroxylation and Oxidation of [closo-B12H12]2– to the Redox System [B12(OH)12]2–/.–: Synthesis, Experimental and Calculated Structures and Properties”, Chem. Eur. J. 2010, 16, 11242–11245.
DOI: 10.1002/chem.201001374.
15. R. S. Chauhan, G. Kedarnath, A. Wadawale, A. Muñoz-Castro, R. Arratia-Perez, V. K. Jain, W. Kaim “Tellurium(0) as a Ligand: Synthesis and Characterization of 2-Pyridyltellurolates of Platinum(II) and Structures of [Pt{2-Te-3-(R)C5H3N}2Te(PR03)] (R = H or Me)”, Inorg. Chem., 2010, 49, 4179-4185.
DOI: 10.1021/ic902347s
14. A. Muñoz-Castro, D. Mac-Leod Carey, and R. Arratia-Perez “Spin-orbit effects on electronic delocalization. Aromaticity in a discrete square tetrapalladium sandwich complex”, J. Chem. Phys. 2010, 132, 164308.
DOI:10.1063/1.3382340.
13. D. Mac-Leod Carey, C. Morales-Verdejo, A. Muñoz-Castro, F. Burgos, D. Abril, C. Adams, E. Molins, O. Cador, I. Chavez, J.M. Manriquez, R. Arratia-Perez, J.Y. Saillard “[Cp*Ru(s-indacene)RuCp*] and [Cp*Ru(s-indacene)RuCp*]+: Experimental and theoretical findings concerning the electronic structure of neutral and mixed valence organometallic systems”, Polyhedron, 2010, 29, 1137-1143.
DOI: 10.1016/j.poly.2009.12.002
12. A. Muñoz-Castro, D. Mac-Leod Carey, R. Arratia-Perez “Calculated Molecular Properties of Triangular Tribenzo and Perfluoro-Tribenzo Trimercuronin Macrocycles”, J. Phys. Chem. A, 2010, 114, 666-672.
DOI:10.1021/jp908493r.
11. A. Muñoz-Castro, D. Mac-Leod Carey, R. Arratia-Pérez “Inside a Superatom: The M7q (M=Cu, Ag, q=1+, 0, 1-) Case”, ChemPhysChem, 2010, 11, 646-650.
10. A. Muñoz-Castro, D. Mac-Leod Carey, C. Morales-Verdejo, I. Chavez, J. M. Manriquez, R. Arratia-Perez “Toward the Synthetic Control of the HOMO-LUMO Gap in Binuclear Systems: Insights from Density Functional Calculations”, Inorg. Chem., 2010, 49, 4175-4178.
DOI: 10.1021/ic902326y.
9. X. Tu, E. Boroson, H. Truong, A. Muñoz-Castro, R. Arratia-Pérez, G.-S. Nichol, Z. Zheng, “Cluster-bound Nitriles Do Not Click with Organic Azides – Unexpected Formation of Imino Complexes of the [Re6(µ3-Se)8]2+ Core-containing Clusters”, Inorg. Chem. 2010, 49, 380-382.
DOI: 10.1021/ic902251z.
8. A. Muñoz-Castro, R. Arratia-Perez “Electronic Delocalization, Energetics, and Optical Properties of Tripalladium Ditropylium Halides, [Pd3(C7H7)2X3]1- (X = Cl-, Br-, and I-)”, J. Phys. Chem. A, 2010, 114, 5217-5221. DOI: 10.1021/jp101038u.
7. A. Muñoz-Castro, D. Mac-Leod Carey, R. Arratia-Perez “Relativistic electronic structure of cadmium(II) multidecker phthalocyanine compounds”, Polyhedron, 2010, 29, 451-455, (2009 Young Investigator Special Issue)
DOI: 10.1016/j.poly.2009.06.038
6. A. Muñoz-Castro, D. Mac-Leod Carey, R. Arratia-Perez “Electronic structure, molecular properties and electronic currents of the luminescent [Au3(CH3NCOCH3)3] cluster”, Chemical Physics Letters, 2009, 474, 290-293,
DOI: 10.1016/j.cplett.2009.04.068
5. A. Muñoz-Castro, D. Mac-Leod Carey, R. Arratia-Pérez, “Electronic structure and molecular properties of binuclear group VII pentalene metal carbonyl complexes [C8H6{M(CO3)}2] (M = Mn, Tc, Re, Bh): A relativistic density functional theory study”, Polyhedron, 2009, 28, 2009, 1561-1567.
DOI: 10.1016/j.poly.2009.03.016
4. J. Hurtado, D. Mac-Leod Carey, A. Muñoz-Castro, R. Arratia-Perez, R. Quijada, G. Wu, R. Rojas, M. Valderrama, “Chromium(III) complexes with terdentade 2,6-bis(azolylmethyl)pyridine ligands: Synthesis, structures and catalitic behavior”, J. Organomet. Chem. 2009, 694, 2636-2641.
DOI: 10.1016/j.jorganchem.2009.04.014
3. J. F. Araneda, S. Salinas, D. Mac-Leod Carey, A. Muñoz-Castro, R. Arratia-Perez, C. Adams, I. Chavez, J.M. Manriquez, “Synthesis, Characterization of a New Carbonylated Zirconium Metallocene using a Dichloro-Zirconocene derived from a partially alkylated s-indacene”, J. Chi. Chem. Soc. 2009, 54, 269-273.
DOI: 10.4067/S0717-97072009000300014
2. C. Adams, C. Morales-Verdejo, V. Morales, D. Mac-Leod Carey, J. M. Manríquez, I. Chávez, A. Muñoz-Castro, F. Delpech, A. Castel, H. Gornitzka, M. Rivière-Baudet, P. Rivière, E. Molins, “Heterobinuclear s-Indacene Rhodium Complexes, Synthesis and Characterization”, Eur. J. Inorg. Chem. 2009, 784-791.
1. D. Mac-Leod Carey, A. Muñoz-Castro, C. J. Bustos, J. M. Manríquez and R.Arratia-Pérez,“Pi-Donor/Acceptor Effect on Lindqvist type polyoxomolibdates because of various multiple-bonded nitrogeneous ligands”, J. Phys. Chem. A 111, 6563-6567 (2007).
DOI: 10.1021/jp0727594